¶ 4D打印金属增强双网络颗粒水凝胶
Matteo Hirsch a, Livia D’Onofrio a, Qinghua Guan b, Josie Hughes b, Esther Amstad a
a 软材料实验室,材料研究所,瑞士洛桑联邦理工学院,1015 洛桑,瑞士
b CREATE实验室,机械工程研究所,瑞士洛桑联邦理工学院,1015 洛桑,瑞士
¶ 摘要
软驱动技术的最新发展对具有局部可变成分、弹性高且足够坚硬以产生显著驱动力的材料提出了需求。水凝胶天然对特定刺激具有响应性,但它们通常面临柔韧性与硬度的折中问题,即柔软且灵活的水凝胶通常难以产生较大的驱动力。通过设计双网络水凝胶可以在一定程度上缓解这一问题,但这种设计的复杂工艺使得局部成分的可控变化变得困难。
通过3D打印技术,可以将水凝胶的成分精确控制到100微米的尺度范围内。如果水凝胶被制备成微粒形式,则可以实现广泛的水凝胶3D打印。然而,由此制得的颗粒状水凝胶通常较为柔软。通过引入贯通的水凝胶网络可以强化颗粒状水凝胶,从而形成双网络颗粒水凝胶(DNGH),但这些水凝胶仍然相对脆弱。
本研究引入了可3D打印的金属增强双网络颗粒水凝胶(mrDNGH),其结合了三个看似矛盾的特性:硬度、韧性和工艺可行性。我们的mrDNGH能够承受高达3 MPa的载荷,并表现出高达12 MJ/m³的断裂能,这一性能至少是以往3D打印水凝胶的20倍。我们还利用mrDNGH的不同膨胀程度,成功3D打印了可形变的结构。
¶ 1. 引言
自然界一直是功能材料设计的重要灵感来源[1]。在这一领域的卓越研究为多种天然材料的成分-结构-功能关系提供了良好的理解[2,3]。例如,对某些软生物组织成分的分析表明,自然界通过非共价相互作用实现了自愈和动态应力响应[4–6]。其中一个显著的例子是贻贝的足丝,这是一种无细胞的软负载组织,使贻贝能够牢固地附着在岩石上,甚至能够承受高达40 MPa的剪切应力冲击波[3]。受自然界的启发,软合成材料通过非共价可逆相互作用得到了机械强化[7–9],如主体-客体相互作用[10,11]、疏水相互作用[12,13]、金属配位[14–17]和可逆共价交联剂[18,19]。
尽管合成软材料的机械性能有了显著提升,但自然界在结构与成分之间的精妙平衡仍然无可匹敌。这种机械性能差距的根本原因在于,人造结构通常具有均匀的结构和成分,而与天然材料明确的分层结构和快速变化的成分相比存在显著差异[20–22]。近年来,已有多种策略被用来在不同长度尺度上控制软合成材料的结构[9,23–25],包括定向自组装[26–29]、相分离[30–34]、微流控技术[35,36]和3D打印[37–41]。然而,这些技术在空间分辨率方面的控制能力有限。
为提高人造软材料微观结构的控制水平并扩大可3D打印软材料的种类,一种新型3D打印材料——“颗粒化微凝胶”被引入[36,42–47]。微凝胶是微米级的水凝胶颗粒,当其浓度超过某一临界体积分数时会发生拥塞[24,48]。然而,由于颗粒之间的连接较弱,这些颗粒状水凝胶通常较为柔软。通过引入贯通的水凝胶网络对颗粒状水凝胶进行强化,该网络能够穿插并共价交联这些颗粒[18,49]。但生成的双网络颗粒水凝胶(DNGH)仍然存在断裂应变有限的问题,裂纹倾向于沿颗粒边界传播[50,51]。
本研究提出了一种可3D打印的金属配位双网络颗粒水凝胶(DNGH),其硬度是目前已报道的所有3D打印水凝胶的12倍,其断裂能则是已报道水凝胶的20倍以上。这一性能的实现基于以下方法:通过聚丙烯酸(PAA)微片与多种阳离子增强交联形成微结构,并与贯通的聚丙烯酰胺(PAM)水凝胶进行共价交联。由此生成的DNGH硬度为0.12 MPa,断裂能为0.02 MJ/m³。当贯通网络通过离子强化时,DNGH的断裂能提高了两个数量级以上,高达12 MJ/m³。实验中,DNGH由PAM微片与金属增强的PAA网络相连接制成。
此外,我们展示了这些材料在软驱动中的潜力,通过3D打印可形变的结构进一步验证其应用前景。
¶ 2. 结果与讨论
为增强颗粒状双网络水凝胶(DNGH)的离子强化,我们采用聚丙烯酸(PAA)作为其中一种水凝胶网络的基础材料。PAA是一种聚电解质,可以通过引入至少二价的离子来强化其性能。为使PAA适用于3D打印,我们将其制备为颗粒形式。颗粒可以通过乳液模板法或微流控技术生成。乳液模板法通过乳化生成颗粒,但需要多次清洗以去除油和表面活性剂,从而限制了生产通量并提高了成本。微流控技术尽管具有更高的尺寸控制能力,但同样受限于通量和生产成本。
另一种更高效的方法是通过机械手段将大块水凝胶粉碎成颗粒,例如使用搅拌机、筛网或冷冻研磨。其中,冷冻研磨特别适合于将玻璃化转变温度高于液氮温度的材料破碎为微片。尽管该方法对颗粒形状的控制较差,但它具有高通量和广泛的材料适应性。通过冷冻研磨法,我们将PAA制备成平均直径为45微米的非球形颗粒,并验证了其尺寸与丙烯酸单体含量无关。
在3D打印之前,将这些颗粒浸泡在丙烯酰胺水溶液中,然后进行拥塞操作以便打印。完成3D打印后,利用紫外线照射启动丙烯酰胺的聚合反应,从而在颗粒间形成贯通网络并交联成DNGH。该网络结构不仅增强了颗粒之间的连接强度,还允许在空间上选择性强化颗粒,实现异质强化的DNGH。这一特性拓展了DNGH在承载和形变水凝胶领域的应用潜力。
DNGH的微观结构由颗粒的尺寸、形态、分布和堆积密度决定。为了优化颗粒的尺寸以避免3D打印时喷嘴堵塞,我们对冷冻研磨过程的参数进行了系统研究。研磨周期数与颗粒尺寸密切相关,经过五次研磨后,颗粒的平均直径降至45微米。然而,约20%的颗粒仍有一个方向的尺寸超过100微米,这些较大的颗粒可能造成喷嘴堵塞,从而影响打印质量。
为解决这一问题,我们使用100微米尼龙筛网对研磨后的颗粒进行过滤。过滤后的颗粒平均尺寸降至20微米,分布更加均匀,并且几乎不含尺寸超过100微米的颗粒。然而,即使经过筛选,仍有少量颗粒因其不规则形状在筛网中对齐适当而穿过筛网。这些颗粒的存在对3D打印提出了更高的工艺要求,需要进一步优化颗粒制备和筛选过程以提升材料均一性。
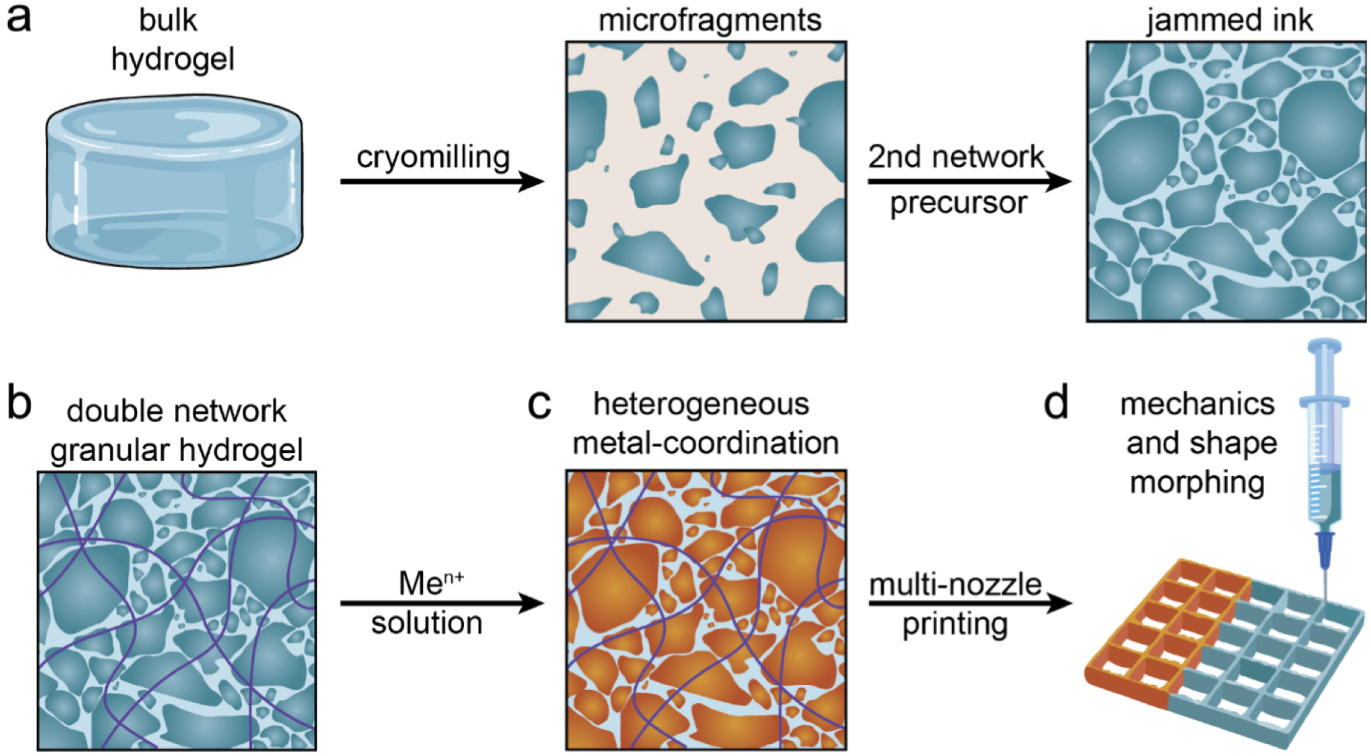
图1. 金属配位双网络颗粒水凝胶(DNGHs)的制备过程
a. 将大块水凝胶进行交联后,通过冷冻研磨方法将其粉碎为多分散的微片。这些获得的微片经过冷冻干燥后,重新分散于含有单体的溶液中,形成适合拥塞操作的3D打印墨水。
b. 拥塞的微片浆料可用于3D打印,并通过紫外线照射将其转化为可承载负载的DNGH结构。
c. 一旦交联完成,将DNGHs浸入含离子的溶液中,以触发微片的机械强化作用。
d. 通过多喷嘴打印技术,可以局部改变DNGHs的成分,实现异质性设计和功能优化。
图2. 通过冷冻研磨制备水凝胶微片的过程
a. 冷冻研磨的示意图。将大块凝胶以30 Hz的频率进行多次冷冻研磨,得到多分散的微片。这些微片随后通过尼龙筛网过滤,去除尺寸大于100微米的颗粒,以避免3D打印过程中喷嘴堵塞的风险。
b. 微片的等效直径(d*)随研磨周期数的变化关系。经过超过5次研磨的微片平均等效直径为45微米。如果继续增加研磨周期数,微片的尺寸和分布没有显著改善。
c. 微片尺寸的累计频率分布随研磨周期数的变化关系。尽管随着研磨周期数的增加,平均等效直径逐渐减小,但约20%的微片仍具有直径大于100微米的颗粒。
d. 经5次研磨后过滤前后的微片等效直径变化。经过过滤,平均等效直径从45微米减少到20微米,并且尺寸分布更加均匀。过滤后,几乎没有尺寸超过100微米的颗粒存在。
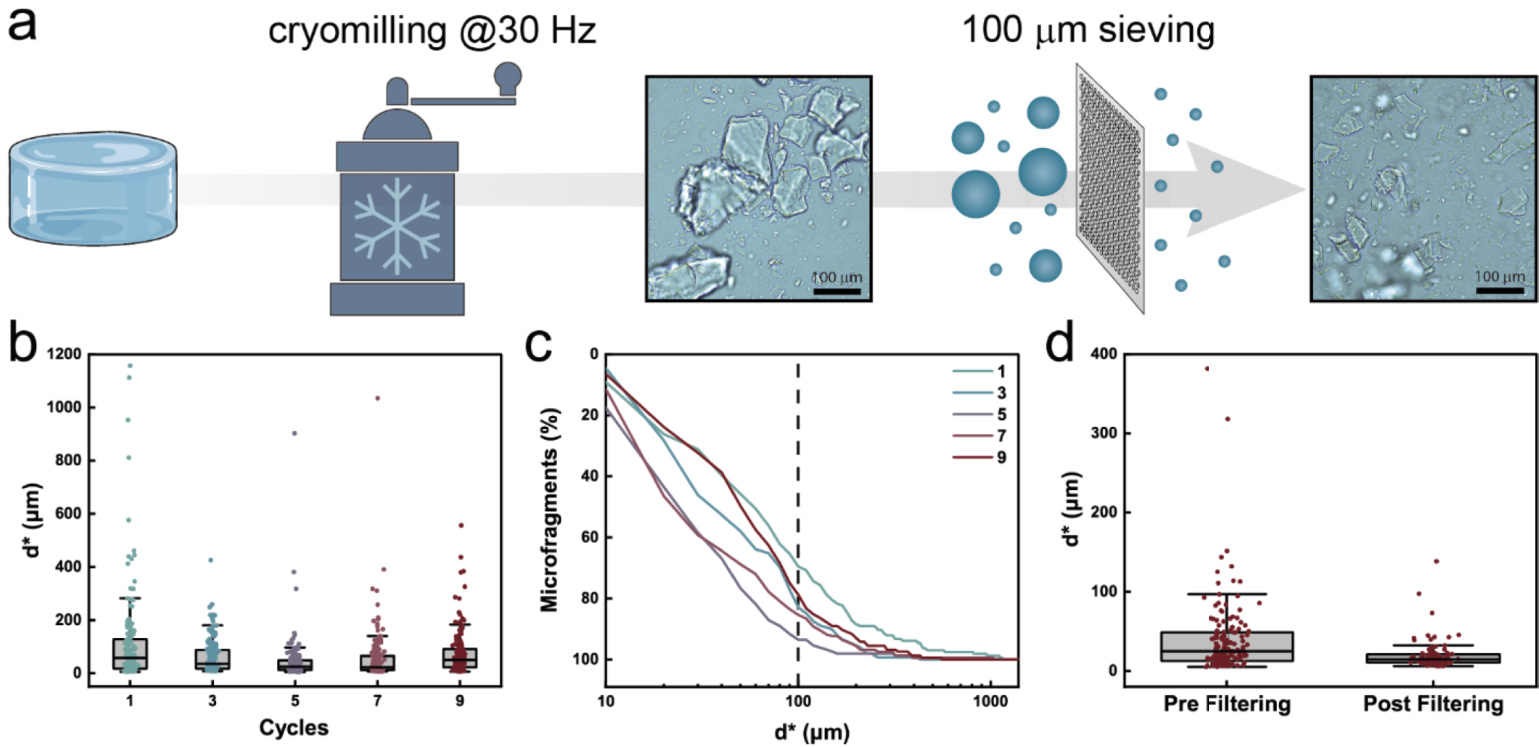
尽管各向异性颗粒可能因剪切对齐而存在,但预期其对3D打印的可打印性影响极小。基于颗粒的多分散性和生产通量的良好结合,选择经过5次冷冻研磨并过滤后的微片作为后续实验的研究材料。这些微片在进一步使用前经过冷冻干燥处理,以延长其储存寿命并更好地控制其拥塞行为。
制备拥塞微片墨水:为制备由拥塞微片组成的墨水,首先将冷冻干燥后的微片重新分散于含有第二种前体溶液的水溶液中。拥塞的软颗粒(包括微凝胶)具有适合挤出式3D打印的流变学特性:低屈服应力、剪切变稀行为以及快速剪切恢复。这些特性使其成为3D打印的理想材料。拥塞微片在流变学行为上与传统的球形微凝胶类似。
流变学性能评估:为评估含反应性单体的拥塞微片系统是否具有类似的流变学特性,对墨水进行了流变学测试。实验中,将墨水中的微片含量按干聚合物重量分数调整在10%至16%之间。通过频率扫描测试微片重量分数变化对剪切变稀行为的影响。结果表明,无论微片含量如何,所有测试样品均表现出剪切变稀行为,显示其流变学性能不受微片含量的显著影响。这些结果证实了拥塞微片系统的流变学性能适合挤出式3D打印(如图3a所示)。
挤出压力与屈服应力的关系:良好的打印性能关键在于低挤出压力,而挤出压力与墨水的屈服应力直接相关。为研究墨水屈服行为与微片含量的关系,进行了振幅扫描测试。随着微片含量从12%增加到16%,墨水的屈服应力从46 Pa增加至2630 Pa(如图3b和图S2a所示)。这一屈服应力的增加被归因于拥塞浆料中聚合物含量的提高,导致微片的拥塞程度加剧。
与传统球形微凝胶的对比:与相同重量分数的球形微凝胶拥塞墨水相比,所有测量墨水的屈服点均更高(如图S2b所示)。这表明微片形态的墨水在屈服应力方面具有更优异的性能,这可能为更高精度和更复杂的3D打印结构提供支持。
屈服点差异的原因:屈服点差异归因于非球形微片的机械嵌锁作用远强于球形微粒的嵌锁作用,从而增加了微片间的颗粒摩擦力。这种增强的机械嵌锁使微片墨水在屈服应力上表现出更高的性能。
快速恢复固态的能力:为了实现高打印精度,墨水在剪切力去除后必须能够快速恢复其固态特性。为评估这一性能,进行了交替剪切恢复循环测试,在1%和100%应变下分别维持200秒。测试结果显示,所有分析的墨水均能快速在固态和液态之间转换,表明其优异的流变学性能(如图3c所示)。
打印精度和分辨率的评估:得益于这些流变学特性,墨水可以以高分辨率3D打印成网格结构。例如,含有16 wt%微片的墨水可打印出细致的网格结构(如图3d所示)。为量化微片墨水的打印分辨率,测量了丝线的横截面积以及网格封闭区域的面积,分析其与微片浓度的关系。结果表明,微片浓度越高,拥塞程度越高,打印分辨率越好(如图3e和3f所示)。
高分辨率与挤出压力的权衡:然而,较高的打印分辨率需要更高的挤出压力(如图3f所示)。由于本研究中的墨水不包含压力敏感组分,因此可以通过使用含16 wt%微片的墨水来最大化打印分辨率。这种优化后的墨水将在后续实验中被广泛应用。
水凝胶用于承载应用的关键特性:水凝胶在承载应用中的关键性能是其在压缩和拉伸条件下承受显著载荷的能力。为了评估这一性能,本研究量化了由聚丙烯酸(PAA)微片和丙烯酰胺网络牢固连接组成的双网络颗粒水凝胶(DNGHs)的刚度和断裂功。
DNGH的制备与机械强化:为将相对脆弱的微片基结构转化为能够承载显著载荷的机械稳定的DNGH,首先将微片加工成适当形状,然后通过紫外光照射引发前体单体的自由基聚合反应。制备完成的DNGH随后被浸泡在去离子水中,直至达到溶胀平衡。
力学性能:制备的DNGH刚度为0.16 MPa,断裂功为0.05 MJ/m³,表明该材料相对柔软。这些结果与之前关于由球形微凝胶组成的DNGH的文献报道一致,证明本研究中DNGH的力学性能符合预期水平。
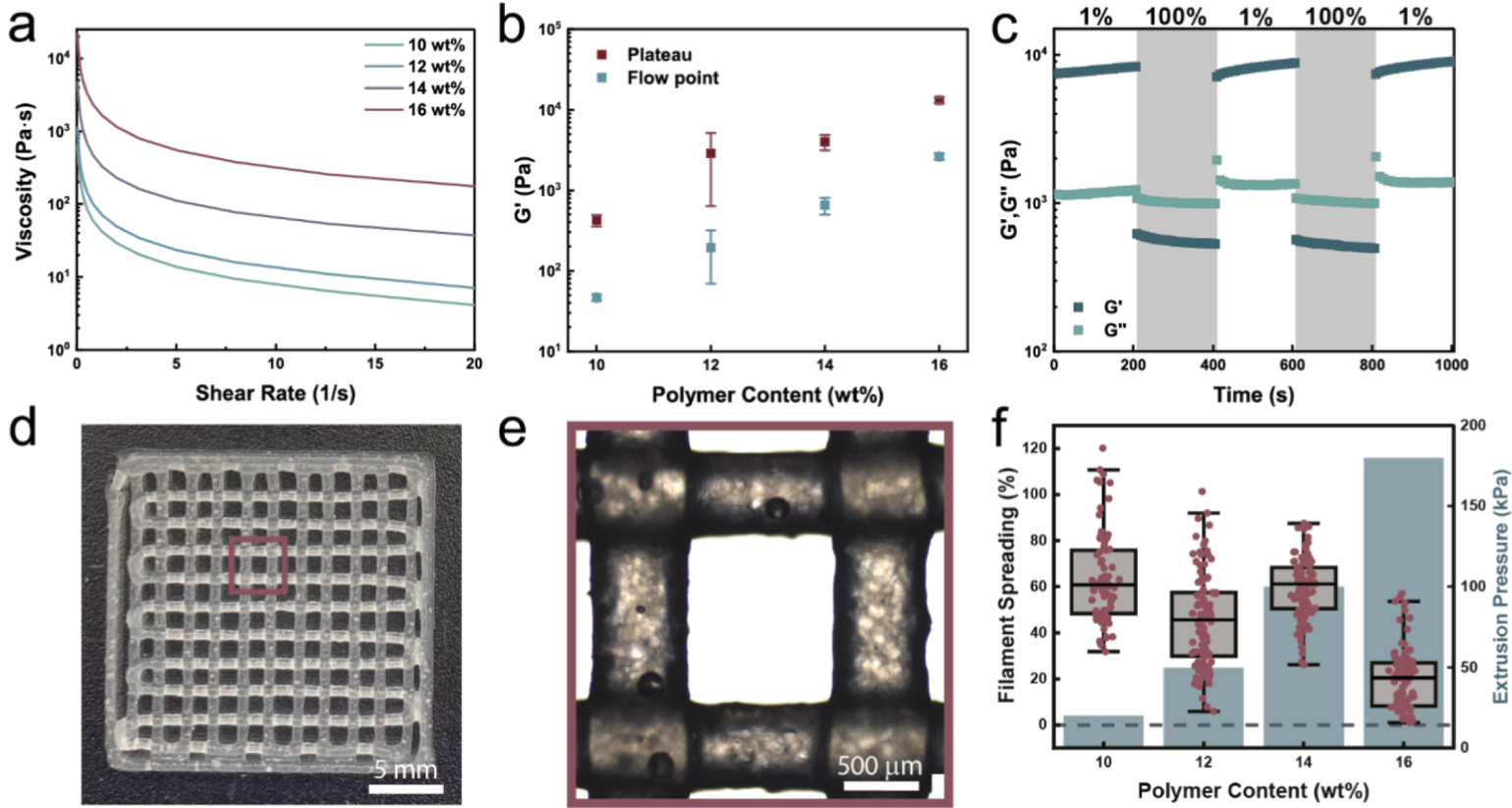
图3. 拥塞微片的可打印性
a. 墨水的频率扫描结果显示,随着剪切速率的增加,所有样品的粘度降低,表现出剪切变稀行为。
b. 墨水的储能模量(G',紫色)和流动点(红色)随微片含量的变化。
c. 剪切恢复测试显示,含有16 wt%聚丙烯酸(PAA)微片的墨水具有自愈行为。在高剪切(γ=100%)下,材料从固态转变为液态;而在低剪切(γ=1%)下,拥塞溶液迅速恢复到初始的固态状态。该过程可多次循环且不会导致墨水性能退化。
d. 光学显微图显示了使用含16 wt%微片的墨水3D打印的网格结构。
e. 网格单元的光学显微图,清晰展示了网格的局部细节。
f. 丝线扩展(红色)和挤出压力(灰色)随墨水中聚合物含量的变化关系。墨水中微片含量的增加与丝线扩展的减少和挤出压力的增加相关联。
离子强化对DNGH断裂功的影响
大块水凝胶在离子强化后通常会表现出更高的断裂功。这是因为聚丙烯酸(PAA)对某些阳离子具有很高的亲和力,可通过离子强化提高机械性能。为验证离子强化是否也能提升DNGH的断裂功,我们将DNGH暴露于含AI3+强化后的微片增强DNGH(mfDNGH)在压缩条件下表现出压缩模量的10倍提升,最高可达1.5 MPa(见图4a和图S3)。在拉伸条件下,杨氏模量提高了25倍,达到4 MPa(见图S4a)。如果改用Fe3+$进行强化,压缩模量和拉伸模量分别提高到25倍和50倍,进一步提升了材料的机械性能(见图4a和图S4a)。
断裂功的提升
经过强化的mfDNGH,其断裂功相比未强化的DNGH增加了16倍,达到0.8 MJ/m³(见图S4b)。这种显著的机械性能提升表明离子强化对于DNGH的韧性和强度具有重要作用。
双价和单价离子的特殊行为
对于含羧基配体并通过适当阳离子强化的水凝胶,通常可以实现超弹性和抗疲劳性能。然而,实验发现,当使用双价离子(如Ca2+和$ Zn2+对mfDNGH进行强化时,机械性能并未显著提升(见图4a)。这一结果与文献中报道的PAA金属强化水凝胶的行为不同。这种现象可能归因于DNGH中羧基(-COOH)和氨基(-NH2)之间更强的配位作用,阻碍了微片与双价离子的进一步复合。
同样地,单价离子(如Ag+对DNGH也未表现出明显的强化效果。这表明,对于不同价态的离子,DNGH的机械性能提升具有较高的选择性,可能需要更深入的配位化学研究以解释这一现象。
均匀金属强化带电大块水凝胶的难点
带电大块水凝胶的均匀金属强化很难实现,因为金属离子的扩散受到金属离子与带电水凝胶之间静电吸引力的限制。这种静电吸引力会导致在水凝胶表面快速形成一个较致密的金属配位水凝胶外壳,从而减缓金属离子在水凝胶内部的扩散。此外,已配位的金属离子会静电排斥自由金属离子,进一步阻碍它们向水凝胶内部渗透。因此,离子强化的大块水凝胶内部通常会出现离子浓度梯度,削弱其机械性能。
通过弱配体复合增强强化
这一限制至少可以部分通过与弱配体复合金属离子来克服。这些配体可以进行竞争性配体交换,从而减弱离子与水凝胶之间的吸引力。为了验证通过弱配体复合是否可以提高离子强化的程度,我们将DNGHs浸泡在含有三氯化铁()和不同浓度柠檬酸(CA)的水溶液中。
实验观察
令人意外的是,随着柠檬酸浓度的增加,mfDNGHs的压缩模量降低(如图4b和图S5所示,结果总结见图4c)。在单轴拉伸负载下也观察到了类似的趋势(见图S6)。这一现象与之前报道的金属强化大块水凝胶形成了鲜明对比。文献中通常认为柠檬酸能够促进离子的均匀分布,从而改善这些水凝胶的整体机械性能。
机制解释
这种现象归因于DNGHs的颗粒结构:
离子在不带电的PAM网络中的扩散比在带电的PAA网络中更加容易,因为缺少或几乎没有静电吸引力。
DNGHs的微片之间仅由PAM网络组成,而微片内部同时存在PAA和PAM网络。因此,离子更容易在PAM单网络中扩散,也更容易在微片之间的区域移动。
在带电的微片内部,扩散路径较短,使得离子能够在不需要竞争性配体交换的情况下均匀强化尺寸达1厘米的mfDNGHs。

图4. mfDNGHs的机械性能表征
a. mfDNGHs的压缩模量随浸泡溶液中离子浓度的变化。当微片通过Fe3+和Al3+强化时,mfDNGHs的刚度显著增加。相比之下,使用单价离子(如Ag+)或双价离子(如Ca2+和Zn2+)强化时,mfDNGHs的刚度并未发生显著变化。
b. 微片通过Fe3+强化的DNGHs的压缩曲线,随柠檬酸(CA)浓度的变化而改变。CA浓度的增加与刚度的降低相关联。
c. 微片通过Fe3+强化的DNGHs的压缩模量随CA浓度的变化。随着CA浓度的增加,压缩模量显著降低。
d. 微片通过Fe3+重复强化并通过CA削弱的DNGHs的压缩模量表现。结果表明,反复强化和削弱对DNGHs的机械性能具有一定影响。
竞争性配体交换在mfDNGHs中的作用
竞争性配体交换通常用于机械强化水凝胶。然而,在本研究系统中,这种方法并非必要。但通过竞争性配体交换,我们仍然可以选择性地移除配位的离子,从而恢复DNGHs的初始机械性能,正如图4d所示。
实验显示,mfDNGHs可以通过将其浸泡在含Fe3+的溶液中进行离子强化,并通过将其暴露于含柠檬酸(CA)的溶液中削弱,以恢复到初始状态。这一过程可以多次循环进行。在第一次循环后,mfDNGHs的刚度略有下降,但在后续的四次强化和削弱循环中,其刚度保持在实验误差范围内,未出现明显变化(如图4d所示)。
金属配位与竞争性配体交换的结合为精确调控mfDNGHs的机械性能提供了可能。这种方法显著扩展了mfDNGHs在软驱动领域的潜在应用,为设计具有动态调控特性的软材料提供了新思路。
我们的研究结果表明,DNGHs(双网络颗粒水凝胶)中微碎片的金属增强显著提高了其刚性。然而,这种增强对断裂功的影响有限。对于体块状双网络水凝胶,其断裂功通常主要由第二网络决定[78,79]。类似地,对于仅通过共价交联的双网络颗粒水凝胶,其断裂功主要由第二网络决定,因为裂纹倾向于优先传播在微凝胶的间隙空间[50]。为了验证是否可以通过离子增强基质网络(即系统中的第二网络)来提高DNGHs的断裂功,我们制备了由未带电的PAM(聚丙烯酰胺)组成的微碎片,并将其浸泡在含有丙烯酸的水溶液中。这些微碎片通过用于制备mfDNGHs的既定方案被加工成基质增强的双网络颗粒水凝胶(mxDNGHs),如图5a所示,并在实验部分中详细描述。
当基质用Al3+增强时,DNGHs的压缩模量增加了8倍;当基质用Fe3+增强时,压缩模量增加了4倍(如图5b所示)。同样,在拉伸条件下测得的杨氏模量在基质用Al3+增强时达到9 MPa,相较于未增强的DNGHs,这对应于75倍的提高;当用Fe3+增强时,则达到略低的3 MPa(如图S7所示)。我们将Al3+增强的DNGHs比Fe3+增强的刚性更高归因于Al3+的结合速度更快,从而导致更刚性的离子增强壳。这些结果暗示mxDNGHs中存在不均匀的离子分布。
为了验证mxDNGHs中的离子浓度是否确实是不均匀的,我们对基质用Fe3+增强的DNGH的截面进行了可视化观察。确实,我们在mxDNGHs的截面上观察到了颜色梯度,与mfDNGHs形成鲜明对比,如图5c所示。这种颜色梯度表明存在核心-壳层结构。通过基质用Fe3+或Al3+增强的DNGHs的压缩曲线进一步支持了这一假设,曲线显示出两个明显不同的斜率,如图5d和图S8所示。
在体块状水凝胶中,可以通过使用柠檬酸(CA)络合离子来最小化离子浓度的梯度,因为这种络合作用减少了离子与水凝胶之间的吸引相互作用,从而促进了离子向水凝胶中的扩散[15,76,77]。为了验证在具有PAA(聚丙烯酸)基质的DNGHs中是否也适用这一机制,我们将其浸泡在含有Fe3+和不同浓度CA的水溶液中。当样品浸泡在含有0.5 M Fe3+和0.75 M CA的溶液中时,mxDNGHs呈现出均匀的颜色。这些结果表明,Fe3+与CA的络合作用提高了mxDNGHs内部Fe3+离子的均匀性。金属增强引入了一些样品间的差异性,这导致了较大的误差范围。我们将这种差异归因于离子在具有微小结构差异的样品中的扩散差异。
尽管如此,我们观察到随着浸泡溶液中CA浓度的增加,压缩模量显著增加,直到达到3 MPa,如图5e所示。在拉伸测试中也观察到了类似的行为,其中mxDNGHs的刚性随着离子溶液中CA浓度的增加而提高,直到达到高达25 MPa的杨氏模量,如图S10所示。有趣的是,当[CA]:[Fe3+]摩尔比超过0.5时,杨氏模量开始下降。这种现象归因于CA溶液中过量的羧基相对于PAA网络中的羧基,使得络合在PAA基质内的Fe3+离子数量减少。
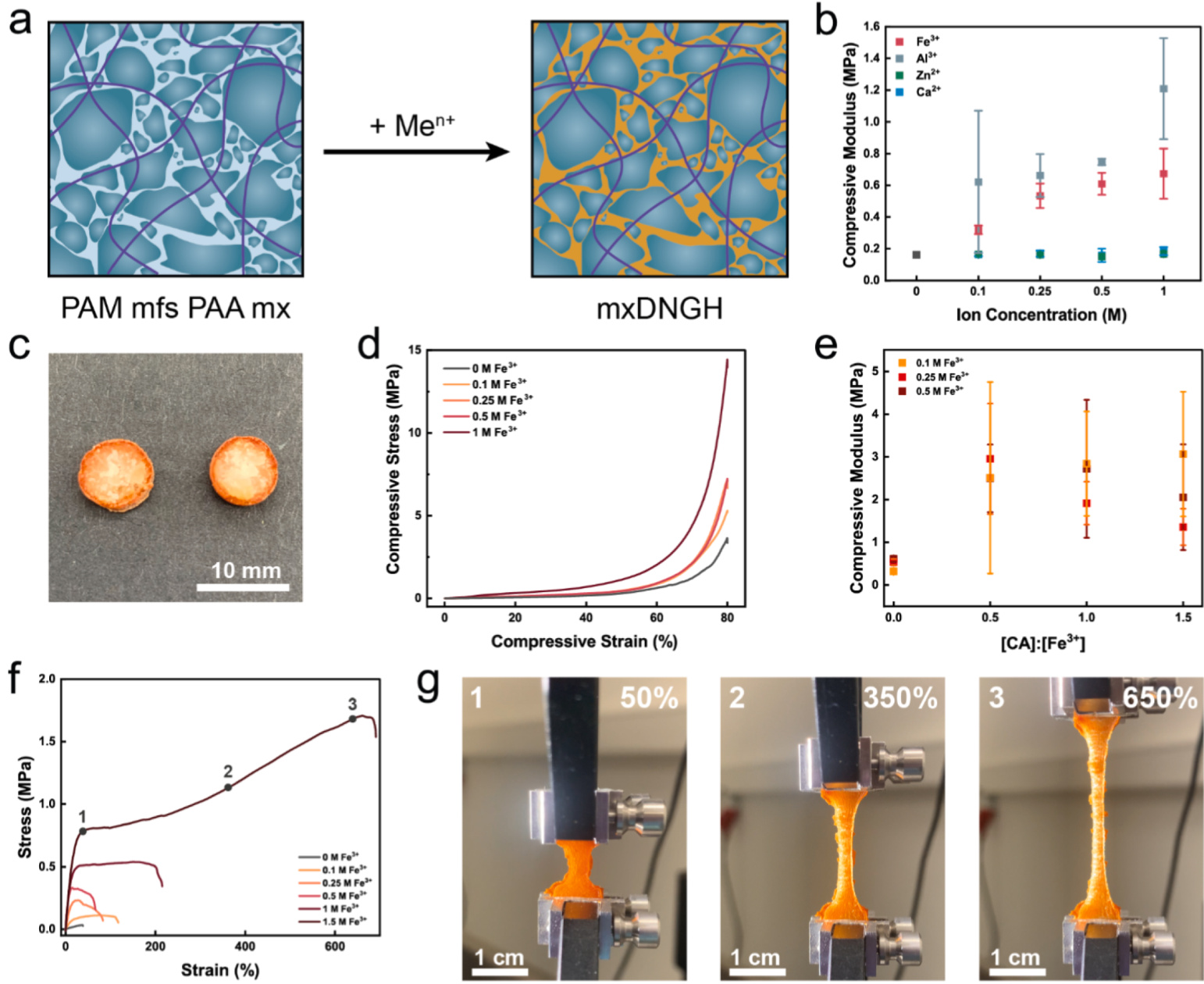
图5. mxDNGHs的机械性能表征。
a. DNGH由PAM微碎片和PAA基质组成。基质通过选择性离子增强形成mxDNGH。
b. mxDNGHs的压缩模量随增强溶液中离子浓度的变化而变化。Fe3+和Al3+显著提高了mxDNGH的刚性,而基质用Ca2+、Zn2+和Ag+增强的DNGHs则未观察到明显变化。
c. mxDNGH圆柱体的光学照片。颜色梯度清晰表明离子增强不均匀。
d. 基质用Fe3+增强的DNGHs的压缩曲线随增强溶液中Fe3+浓度的变化而变化。所有曲线都表现出初始的柔软行为,随后刚性增加。这种刚性变化表明存在核心-壳层结构。
e. 基质用Fe3+增强的mxDNGHs的压缩模量随CA浓度的变化而变化。CA浓度的增加伴随着压缩模量的显著提升。CA减缓了离子络合作用,从而产生更均匀、更坚固的结构。
f. mxDNGHs的拉伸曲线随增强溶液中Fe3+浓度的变化而变化。更高的Fe3+浓度使mxDNGHs的屈服平台更加明显,这表明形成了核心-壳层结构。
g. 基质用Fe3+增强的mxDNGH的时间序列图。最初样品显示出均匀的纹理(1)。当mxDNGH拉伸超过其屈服点时,硬壳发生裂纹(2),并显现出更柔软且具有弹性的核心(3)。
刚性增加的同时,断裂应变显著降低,如图S11所示。断裂应变的降低通常会减少软材料的韧性,使其变得更加脆弱。为了验证这种现象是否也适用于我们的系统,我们通过计算应力-应变曲线下的面积,提取了断裂功与[CA]:[Fe3+]摩尔比的关系。有趣的是,断裂功几乎保持不变,约为0.20 MJ·m⁻³。该结果与之前关于金属增强体块状水凝胶的文献一致,表明此类系统的潜力[15]。
金属配位通过在现有的共价网络中引入物理交联点,提高了双交联水凝胶的刚性和韧性[72]。然而,交联密度的增加以牺牲水含量为代价:在金属增强过程中,水凝胶通常会发生收缩(聚沉),导致其体积缩小。为了评估我们的mrDNGHs在多大程度上发生了聚沉,我们通过重力分析,量化了所有样品的水含量与增强溶液中离子浓度的关系。如预期的那样,mrDNGHs中离子浓度的增加伴随着水含量的降低,当mxDNGHs暴露于含1.5 M AlCl₃的水溶液中时,水含量降至最低约24 wt%,如图S12所示。
我们的研究结果表明,离子交联密度的增加与这些水凝胶中水含量的减少直接相关。尽管这种行为对细胞工作可能有不利影响,但在制造承重型高韧性水凝胶时却是一种优势。事实上,金属离子引发的二次交联导致的收缩迫使聚合物链塌缩,从而增加了其隐藏长度,提高了水凝胶的断裂韧性[9]。自然界经常利用这种行为构建具有无与伦比韧性的基于水凝胶的材料[2,6,80]。
为了验证我们是否能够在mxDNGHs中促进这种行为,我们在没有CA的情况下用Fe3+对其进行增强,以形成收缩的刚性壳,从而机械地迫使壳内的核心也发生塌缩。确实,我们在用Fe3+浓度≥1 M溶液增强的样品的应力-应变曲线中观察到了两种明显不同的斜率,如图5f和图5g所示。这些DNGHs的应力-应变曲线中两种明显不同的斜率,反映了两种截然不同的刚性,表明它们具有核心-壳层结构:当基质用高浓度Fe3+增强时,这些DNGHs在拉伸下表现出初始的刚性行为,我们将其归因于刚性Fe3+增强的壳层。这种应力-应变曲线中最初的陡增随后出现一个明显的屈服平台,表明核心部分更柔软,这一现象在拉伸测试中的样品变形视频(Movie M1)中得到了验证。当DNGHs的基质用Al3+增强时,也观察到了类似的行为,如图S13所示。
总体而言,随着增强溶液中离子浓度的增加,mxDNGHs的断裂强度和断裂应变均有所提高,最终达到高达9 MPa的刚性和12 MJ·m⁻³的断裂功,如图S7和图S9所示。
为了评估mfDNGHs和mxDNGHs的整体机械性能,我们将其与当前最先进的3D打印水凝胶进行了比较。值得注意的是,我们的材料在刚性和断裂功方面的数值高于任何已报道的3D打印水凝胶,如图6a所示。为了评估这些优异的机械性能是否仅与高聚合物含量相关,我们将我们的配方与之前报道的3D打印水凝胶按聚合物含量进行了比较。即使将我们的材料与具有相似聚合物含量的水凝胶进行比较,其刚性和韧性仍显著高于任何之前报道的3D打印水凝胶,如图6b和图6c所示。这些结果突出了我们的3D可打印水凝胶在承重应用中的潜力。
为了展示mfDNGHs的3D打印技术与金属配位的协同作用,我们打印了一个铰链(如图7a所示),并用Fe3+进行增强(如图7b所示)。增强后的铰链,其截面积为0.36 cm²,可以承受高达1.5 kg的重量且无明显损伤(如图7c所示)。这一例子表明,3D打印的mfDNGHs在作为承重结构方面具有巨大的潜力。
通过这些测试,我们不仅证明了mfDNGHs和mxDNGHs在机械性能上的卓越表现,还展示了其在实际应用中的可操作性和适用性。这种材料结合了3D打印的复杂结构制造能力和金属配位的机械性能增强特性,为开发新型高性能水凝胶材料提供了强有力的支持。尤其是在负载应用、生物医学工程以及软体机器人领域,这种材料可能具有广泛的应用前景。
我们的研究结果表明,基质的金属配位增加了水凝胶的交联密度,从而排出了大量的水,导致mrDNGHs发生收缩。我们利用这种体积变化来实现4D打印形状变化结构:通过将3D打印的具有空间上不同成分的结构暴露于含有增强Fe3+离子的水溶液中,触发其形状改变。
我们制造了由PAA微碎片组成的区域和由惰性PAM微碎片组成的区域的结构,后者不能通过离子交联,因此在含Fe3+的溶液中不会收缩(如图7d和图7e所示)。确实,在暴露于含FeCl₃的溶液后,由PAA微碎片组成的区域由于结构的离子增强显著收缩。相比之下,由PAM微碎片组成的区域体积没有显著变化。由于这种溶胀行为的差异,我们成功触发了打印结构的形状变化,如图7f和图7g所示。这种形状变化与通过Abaqus模拟的行为高度一致,如图7h和图7i所示。
这表明,通过合理设计材料的空间分布和成分,可以利用4D打印技术实现复杂的形状变化,为动态可调材料和结构的开发提供了新的思路和可能性。
- 结论
我们介绍了一种可3D打印的金属增强双网络颗粒水凝胶(DNGHs),其刚性和韧性至少是以往报道的3D打印水凝胶的9倍以上。通过选择合适的增强离子,可以将这些DNGHs的机械性能调整为类似于某些坚硬且韧性的天然高分子材料,例如软骨和肌腱。通过低温研磨制备微碎片的技术具有极大的潜力,可用于大规模生产材料,得益于其良好的可扩展性以及处理多种材料的能力。我们利用这种技术在材料选择上的灵活性,使水凝胶的特定区域能够对金属离子产生响应。金属离子对PAA的选择性结合实现了3D打印结构的定向增强,而无需在刚性和韧性之间进行权衡。
我们预计,这些材料良好的加工性能以及高刚性和韧性将为这些水凝胶的应用开辟新领域,其性能显著优于以往报道的3D打印水凝胶。由于其承重性能,我们展望这些材料可用于制造下一代承重软体机器人,使其能够实现比现有技术更复杂的运动。通过进一步优化其成分以实现生物相容性,我们甚至可以预见这些材料在组织替代领域中的应用前景。
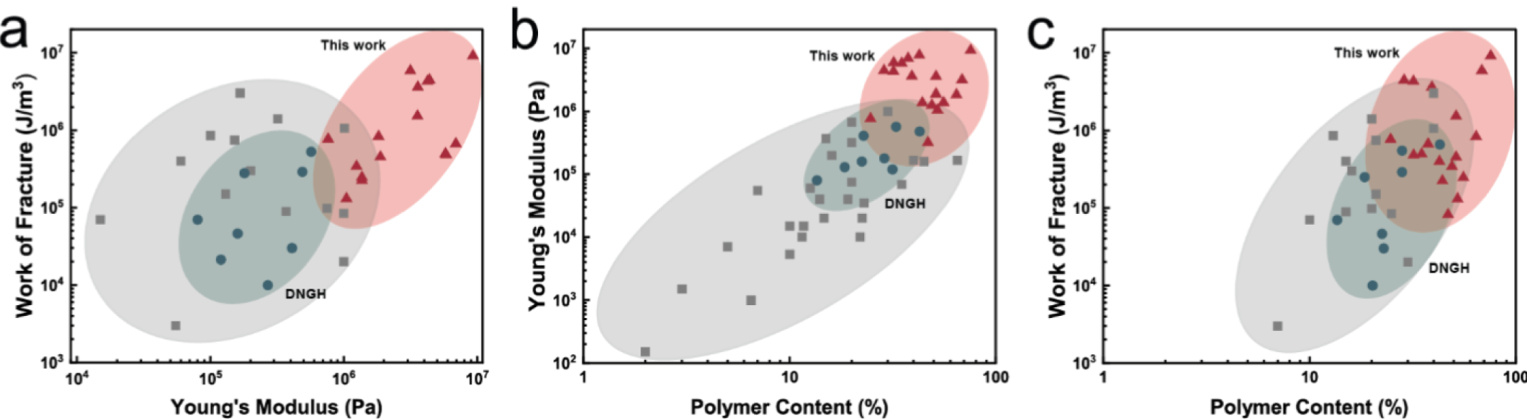
图6. 3D打印水凝胶的Ashby图。
a. 3D打印水凝胶的断裂功与其杨氏模量的Ashby图。我们的材料展现出高刚性和高断裂功的协同组合,这一特性在通常情况下是相互对立的。
b. 3D打印水凝胶的杨氏模量与其聚合物含量的Ashby图。mfDNGHs和mxDNGHs的刚性明显高于任何已报道的3D打印水凝胶。
c. 3D打印水凝胶的断裂功与其聚合物含量的Ashby图。mfDNGHs和mxDNGHs的断裂功显著高于任何已报道的3D打印水凝胶。
灰色阴影区域表示当前最先进的承重3D打印水凝胶的力学性能;绿色阴影区域代表未增强DNGHs的力学性能;红色阴影区域总结了mfDNGHs和mxDNGHs的力学性能。
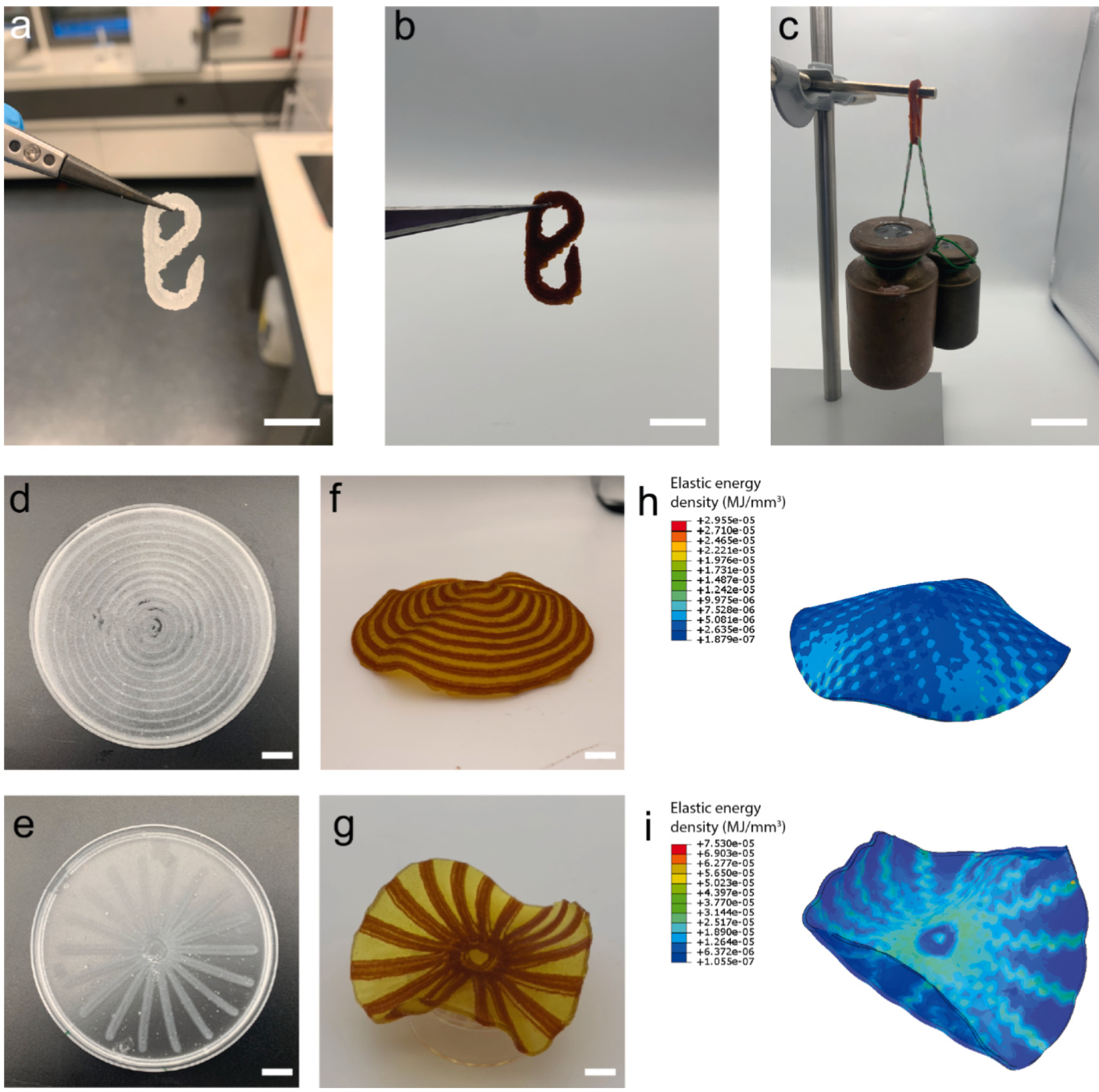
图7. 3D打印mfDNGHs的应用。
a,b. 3D打印的mfDNGH铰链在增强前(a)和用Fe3+增强后(b)的照片。比例尺为2 cm。
c. mfDNGH铰链承受1.5 kg重量的照片。比例尺为5 cm。
d-i. 4D打印形状变形的mfDNGHs。
(d,e) 由用Fe3+增强的DNGHs微碎片(红色)和未被离子增强的微碎片(黄色)组成的3D打印DNGHs的照片。
(f,g) 形状变形结构的照片。
(h,i) 对应的Abaqus模拟。比例尺为1 cm。
- 方法
4.1 材料
以下化学试剂均直接使用:丙烯酸 (AA) (Sigma-Aldrich, 147230)、丙烯酰胺 (AM) (Sigma-Aldrich, A4058)、N,N’-亚甲基双丙烯酰胺 (MBA) (Carl Roth, 7867.1)、2-羟基-2-甲基苯丙酮 (PI) (Sigma-Aldrich, 405655)、轻质矿物油 (Sigma-Aldrich, 330779)、Span80 (TCI Chemicals, S0060)、氯化铁 (III) (Sigma-Aldrich, 451649)、氯化铝 (Sigma-Aldrich, 563919)、氯化钙 (Sigma-Aldrich, 499609)、氯化锌 (Sigma-Aldrich, 208086)、硝酸银 (Sigma-Aldrich, 209139)、柠檬酸 (Sigma-Aldrich, 251275)、乙醇 (Sigma-Aldrich, 459844)。
4.2 PAA和PAM水凝胶的制备
配制含30 wt%单体(AA或AM)、1 wt% MBA交联剂以及5 mg/mL PI的水溶液。将溶液倒入直径为20 cm的培养皿中,并在UV烘箱(UVChamber-365-100, UWave, 25 mW/cm²)中聚合5分钟,得到体块状水凝胶。
4.3 微碎片的制备
将体块状水凝胶用商用搅拌机破碎后分散在水中,直至达到溶胀平衡。随后将得到的水凝胶碎片在液氮(LN2)中冷冻,再用振动冷冻研磨机(Cryomill, Retsch)及12个直径为10 mm的不锈钢研磨球进行冷冻研磨。冷冻研磨的具体操作步骤如下:预冷3分钟;随后进行5个研磨周期,每周期3分钟,频率为30 Hz;每个周期之间间隔30秒,频率为5 Hz,除非有特殊说明。
研磨后的微碎片悬浮于水中,并使用100 μm网孔的尼龙滤网过滤以去除较大的碎片。最后,将微碎片冻干并以粉末形式储存备用。
4.4 微凝胶的制备
PAA微凝胶按照文献[49]中的方法制备。简要来说,配制含30 wt%丙烯酸(AA)、1 wt% MBA交联剂和5 mg/mL PI的水溶液。将此水相以5:1的体积比乳化到含2 wt% Span80的矿物油基溶液中。水包油乳液在搅拌下暴露于UV光(OmniCure S1000, Lumen Dynamics, 320-390 nm, 60 mW/cm²)照射5分钟,将液滴转化为微凝胶。制得的PAA微凝胶转移到乙醇中并以4500 rpm离心10分钟(Mega Star 1.6R, VWR),以去除油相。弃去上清液后,用乙醇重复此过程三次,然后用水重复三次。清洗后的PAA微凝胶悬浮于水中保存。
4.5 微碎片的流变学表征
流变学测试在DHR-3 TA仪器上进行,采用直径为8 mm的平行钢板几何。所有测试均在25°C下进行,间隙设为800 μm。每次测试前,样品静置200秒。频率相关的粘度测试在0.5%应变下进行。振幅扫描在1.0 rad/s的振荡频率下进行。自愈性能测试在1.0 rad/s下进行,交替施加200秒1%应变和200秒100%应变。
4.6 微碎片的3D打印
将堵塞状态的微碎片膏体装入一个3 mL Luer锁口注射器中。注射器密封后,以4500 rpm离心5分钟,以去除可能影响打印质量的气泡。3D打印使用商业3D生物打印机(BIO X, Cellink),打印速度为10 mm/s,压力为100 kPa,除非另有说明。颗粒墨水通过一个锥形喷嘴(22G,内径0.41 mm)在压力驱动的活塞作用下挤出。
4.7 mfDNGH的制备
将PAA微碎片悬浮于含有30%丙烯酰胺(AM)、0.5 wt% MBA和5 mg/mL PI的水溶液中。将微碎片以4500 rpm离心10分钟后弃去上清液,得到堵塞状态的膏体。将膏体倒入狗骨形模具(横截面为5×2 mm²)或圆柱形模具(直径5 mm,高度5 mm)中,并在UV光照下(UVChamber-365-100, UWave, 25 mW/cm²)交联5分钟。
4.8 mxDNGH的制备
将PAM微碎片悬浮在含有30%丙烯酸(AA)、0.5 wt% MBA和5 mg/mL PI的水溶液中,采用与制备mfDNGH相同的步骤进行处理。
4.9 mfDNGH和mxDNGH的机械性能表征
机械测试使用商用仪器(zwickiLine 5 kN,50 N传感器,Zwick Roell)进行。压缩测试在圆柱形样品上进行(直径为8 mm,高度为8 mm),以3 mm/min的恒定速度压缩至断裂或80%应变。压缩模量通过0%-5%应变范围内的斜率计算。
拉伸测试在狗骨形样品上进行(标距截面为5×2 mm²),以100 mm/min的恒定速度拉伸。杨氏模量通过5%-15%应变范围内初始线性区域的斜率计算。断裂功(WOF)通过应力-应变曲线下的面积计算。
4.10 形状变形的模拟
形状变形性能通过有限元建模(FEM)分析,使用Abacus软件对具有空间体积变化的结构进行模拟。形状变形结构中对应于mrDNGHs的区域使用线性梁单元(Abacus Beam Elements B31)表示,未增强的DNGHs区域使用二次四面体单元(Abacus Tetrahedron Elements C3D10)表示。mrDNGHs的区域嵌入在软相中,假设活性材料的收缩线性与施加的载荷相关,扩展比为-0.0016,场载荷范围为1-100。
为了模拟形状变形的演变,活性相和软相初始化为相同的弹性模量,并逐步将增强区域的模量增加15倍。
¶ 参考文献
[1] L. Ng, M.A. Elgar, D. Stuart-Fox, From bioinspired to bioinformed: benefits of greater engagement from biologists, accessed August 15, 2022, Front. Ecol. Evol. 9 (2021), https://www.frontiersin.org/articles/10.3389/fevo.2021.790270.
[2] M.J. Harrington, F. Jehle, T. Priemel, Mussel byssus structure-function and fabrication as inspiration for biotechnological production of advanced materials, Biotechnol. J. 13 (2018) 1800133, https://doi.org/10.1002/biot.201800133.
[3] C.N.Z. Schmitt, Y. Politi, A. Reinecke, M.J. Harrington, Role of sacrificial proteinmetal bond exchange in mussel byssal thread self-healing, Biomacromolecules 16 (2015) 2852–2861, https://doi.org/10.1021/acs.biomac.5b00803.
[4] M.J. Harrington, A. Masic, N. Holten-Andersen, J.H. Waite, P. Fratzl, Iron-clad fibers: a metal-based biological strategy for hard flexible coatings, Science 328 (2010) 216–220, https://doi.org/10.1126/science.1181044.
[5] M. Krogsgaard, V. Nue, H. Birkedal, Mussel-inspired materials: self-healing through coordination chemistry, Chem. – Eur. J. 22 (2016) 844–857, https://doi.org/ 10.1002/chem.201503380.
[6] S. Zechel, M.D. Hager, T. Priemel, M.J. Harrington, Healing through histidine: bioinspired pathways to self-healing polymers via imidazole-metal coordination, Biomimetics. 4 (2019) 20, https://doi.org/10.3390/biomimetics4010020.
[7] A. Charlet, F. Bono, E. Amstad, Mechanical reinforcement of granular hydrogels, Chem. Sci. 13 (2022) 3082–3093, https://doi.org/10.1039/D1SC06231J.
[8] X. Zhao, Multi-scale multi-mechanism design of tough hydrogels: building dissipation into stretchy networks, Soft Matter 10 (2014) 672–687, https://doi. org/10.1039/C3SM52272E.
[9] X. Zhao, X. Chen, H. Yuk, S. Lin, X. Liu, G. Parada, Soft materials by design: unconventional polymer networks give extreme properties, Chem. Rev. 121 (2021) 4309–4372, https://doi.org/10.1021/acs.chemrev.0c01088.
[10] J. Jin, L. Cai, Y.-G. Jia, S. Liu, Y. Chen, L. Ren, Progress in self-healing hydrogels assembled by host– guest interactions: preparation and biomedical applications, J. Mater. Chem. B 7 (2019) 1637–1651, https://doi.org/10.1039/C8TB02547A.
[11] C. Li, M.J. Rowland, Y. Shao, T. Cao, C. Chen, H. Jia, X. Zhou, Z. Yang, O. A. Scherman, D. Liu, Responsive double network hydrogels of interpenetrating dna and cb[8] host-guest supramolecular systems, Adv. Mater. 27 (2015) 3298–3304, https://doi.org/10.1002/adma.201501102.
[12] J. Chen, R. An, L. Han, X. Wang, Y. Zhang, L. Shi, R. Ran, Tough hydrophobic association hydrogels with self-healing and reforming capabilities achieved by polymeric core-shell nanoparticles, Mater. Sci. Eng. C 99 (2019) 460–467, https:// doi.org/10.1016/j.msec.2019.02.005.
[13] L. Xu, X. Zhao, C. Xu, N.A. Kotov, Water-rich biomimetic composites with abiotic self-organizing nanofiber network, Adv. Mater. 30 (2018) 1703343, https://doi. org/10.1002/adma.201703343.
[14] A. Andersen, M. Krogsgaard, H. Birkedal, Mussel-inspired self-healing doublecross-linked hydrogels by controlled combination of metal coordination and covalent cross-linking, Biomacromolecules 19 (2018) 1402–1409, https://doi.org/ 10.1021/acs.biomac.7b01249.
[15] M. Hirsch, M. Steinacher, R. Zhao, E. Amstad, Load-bearing hydrogels ionically reinforced through competitive ligand exchanges, Biomater. Sci. 9 (20) (2021) 6753–6762.
[16] A. Charlet, V. Lutz-Bueno, R. Mezzenga, E. Amstad, Shape retaining self-healing metal-coordinated hydrogels, Nanoscale 13 (7) (2021) 4073–4084.
[17] S.Y. Zheng, H. Ding, J. Qian, J. Yin, Z.L. Wu, Y. Song, Q. Zheng, Metal-coordination complexes mediated physical hydrogels with high toughness, stick-slip tearing behavior, and good processability, Macromolecules 49 (2016) 9637–9646, https:// doi.org/10.1021/acs.macromol.6b02150.
[18] A. Charlet, M. Hirsch, S. Schreiber, E. Amstad, Recycling of load-bearing 3d printable double network granular hydrogels, Small 18 (12) (2022) 2107128.
[19] N. Zheng, Y. Xu, Q. Zhao, T. Xie, Dynamic covalent polymer networks: a molecular platform for designing functions beyond chemical recycling and self-healing, Chem. Rev. 121 (2021) 1716–1745, https://doi.org/10.1021/acs. chemrev.0c00938.
[20] P. Fratzl, R. Weinkamer, Nature’s hierarchical materials, Prog. Mater Sci. 52 (2007) 1263–1334, https://doi.org/10.1016/j.pmatsci.2007.06.001.
[21] C. Chen, Y. Kuang, S. Zhu, I. Burgert, T. Keplinger, A. Gong, T. Li, L. Berglund, S. J. Eichhorn, L. Hu, Structure–property–function relationships of natural and engineered wood, Nat. Rev. Mater. (2020) 1–25, https://doi.org/10.1038/s41578- 020-0195-z.
[22] P. Fratzl, Biomimetic materials research: what can we really learn from nature’s structural materials? J. R. Soc. Interface 4 (2007) 637–642, https://doi.org/ 10.1098/rsif.2007.0218.
[23] A.S. Caldwell, G.T. Campbell, K.M.T. Shekiro, K.S. Anseth, Clickable microgel scaffolds as platforms for 3d cell encapsulation, Adv. Healthc. Mater. 6 (2017) 1700254, https://doi.org/10.1002/adhm.201700254.
[24] A.C. Daly, L. Riley, T. Segura, J.A. Burdick, Hydrogel microparticles for biomedical applications, Nat. Rev. Mater. 5 (2020) 20–43, https://doi.org/10.1038/s41578- 019-0148-6.
[25] J.M. Lowen, J.K. Leach, Functionally graded biomaterials for use as model systems and replacement tissues, Adv. Funct. Mater. 30 (2020) 1909089, https://doi.org/ 10.1002/adfm.201909089.
[26] F. Jehle, P. Fratzl, M.J. Harrington, Metal-tunable self-assembly of hierarchical structure in mussel-inspired peptide films, ACS Nano 12 (2018) 2160–2168, https://doi.org/10.1021/acsnano.7b07905.
[27] H. Tan, C. Xiao, J. Sun, D. Xiong, X. Hu, Biological self-assembly of injectable hydrogel as cell scaffold via specific nucleobase pairing, Chem. Commun. 48 (2012) 10289–10291, https://doi.org/10.1039/C2CC35449G.
[28] R. Blell, X. Lin, T. Lindstr¨om, M. Ankerfors, M. Pauly, O. Felix, G. Decher, Generating in-plane orientational order in multilayer films prepared by sprayassisted layer-by-layer assembly, ACS Nano 11 (2017) 84–94, https://doi.org/ 10.1021/acsnano.6b04191.
[29] W. Li, P. Zhao, C. Lin, X. Wen, E. Katsanevakis, D. Gero, O. F´elix, Y. Liu, Natural polyelectrolyte self-assembled multilayers based on collagen and alginate: stability and cytocompatibility, Biomacromolecules 14 (2013) 2647–2656, https://doi.org/ 10.1021/bm4005063.
[30] S. Cha, H.G. Lim, M.F. Haase, K.J. Stebe, G.Y. Jung, D. Lee, Bicontinuous interfacially jammed emulsion gels (bijels) as media for enabling enzymatic reactive separation of a highly water insoluble substrate, Sci. Rep. 9 (2019) 6363, https://doi.org/10.1038/s41598-019-42769-8.
[31] G. Di Vitantonio, T. Wang, M.F. Haase, K.J. Stebe, D. Lee, Robust bijels for reactive separation via silica-reinforced nanoparticle layers, ACS Nano 13 (2019) 26–31, https://doi.org/10.1021/acsnano.8b05718.
[32] W. Lei, S. Qi, Q. Rong, J. Huang, Y. Xu, R. Fang, K. Liu, L. Jiang, M. Liu, Diffusion–freezing-induced microphase separation for constructing large-area multiscale structures on hydrogel surfaces, Adv. Mater. 31 (2019) 1808217, https://doi.org/10.1002/adma.201808217.
[33] G.-L. Ying, N. Jiang, S. Maharjan, Y.-X. Yin, R.-R. Chai, X. Cao, J.-Z. Yang, A. K. Miri, S. Hassan, Y.S. Zhang, Aqueous two-phase emulsion bioink-enabled 3d bioprinting of porous hydrogels, Adv. Mater. 30 (2018) 1805460, https://doi.org/ 10.1002/adma.201805460.
[34] W. Jia, P.S. Gungor-Ozkerim, Y.S. Zhang, K. Yue, K. Zhu, W. Liu, Q. Pi, B. Byambaa, M.R. Dokmeci, S.R. Shin, A. Khademhosseini, Direct 3D bioprinting of perfusable vascular constructs using a blend bioink, Biomaterials 106 (2016) 58–68, https:// doi.org/10.1016/j.biomaterials.2016.07.038.
[35] M. Kessler, H. Elettro, I. Heimgartner, S. Madasu, K.A. Brakke, F. Gallaire, E. Amstad, Everything in its right place: controlling the local composition of hydrogels using microfluidic traps, Lab Chip 20 (24) (2020) 4572–4581.
[36] H. Du, A. Cont, M. Steinacher, E. Amstad, Fabrication of hexagonal-prismatic granular hydrogel sheets, Langmuir 34 (2018) 3459–3466, https://doi.org/ 10.1021/acs.langmuir.7b04163.
[37] D. Chimene, C.W. Peak, J.L. Gentry, J.K. Carrow, L.M. Cross, E. Mondragon, G. B. Cardoso, R. Kaunas, A.K. Gaharwar, Nanoengineered ionic-covalent entanglement (nice) bioinks for 3d bioprinting, ACS Appl. Mater. Interfaces 10 (2018) 9957–9968, https://doi.org/10.1021/acsami.7b19808.
[38] G. Ying, N. Jiang, C. Parra-Cantu, G. Tang, J. Zhang, H. Wang, S. Chen, N.- P. Huang, J. Xie, Y.S. Zhang, Bioprinted injectable hierarchically porous gelatin methacryloyl hydrogel constructs with shape-memory properties, Adv. Funct. Mater. 30 (2020) 2003740, https://doi.org/10.1002/adfm.202003740.
[39] R. Levato, T. Jungst, R.G. Scheuring, T. Blunk, J. Groll, J. Malda, From shape to function: the next step in bioprinting, Adv. Mater. 32 (2020) 1906423, https://doi. org/10.1002/adma.201906423.
[40] K.S. Lim, R. Levato, P.F. Costa, M.D. Castilho, C.R. Alcala-Orozco, K.M.A. van Dorenmalen, F.P.W. Melchels, D. Gawlitta, G.J. Hooper, J. Malda, T.B. F. Woodfield, Bio-resin for high resolution lithography-based biofabrication of complex cell-laden constructs, Biofabrication 10 (2018), 034101, https://doi.org/ 10.1088/1758-5090/aac00c.
[41] L. Aydin, S. Kucuk, H. Kenar, A universal self-eroding sacrificial bioink that enables bioprinting at room temperature, Polym. Adv. Technol. 31 (7) (2020) 1634–1647.
[42] A.C. Daly, M.D. Davidson, J.A. Burdick, 3D bioprinting of high cell-density heterogeneous tissue models through spheroid fusion within self-healing hydrogels, Nat. Commun. 12 (2021) 753, https://doi.org/10.1038/s41467-021- 21029-2.
[43] J.E. Mealy, J.J. Chung, H.-H. Jeong, D. Issadore, D. Lee, P. Atluri, J.A. Burdick, Injectable granular hydrogels with multifunctional properties for biomedical applications, Adv. Mater. 30 (2018) 1705912, https://doi.org/10.1002/ adma.201705912.
[44] V.G. Muir, T.H. Qazi, J. Shan, J. Groll, J.A. Burdick, Influence of microgel fabrication technique on granular hydrogel properties, ACS Biomater Sci. Eng. 7 (9) (2021) 4269–4281.
[45] T.H. Qazi, J. Wu, V.G. Muir, S. Weintraub, S.E. Gullbrand, D. Lee, D. Issadore, J.A. Burdick, Anisotropic Rod-Shaped Particles Influence Injectable Granular Hydrogel Properties and Cell Invasion, (2021) 2021.09.23.461542. https://doi.org/ 10.1101/2021.09.23.461542.
[46] S. Xin, D. Chimene, J.E. Garza, A.K. Gaharwar, D.L. Alge, Clickable PEG hydrogel microspheres as building blocks for 3D bioprinting, Biomater. Sci. 7 (2019) 1179–1187, https://doi.org/10.1039/C8BM01286E.
[47] C.B. Highley, K.H. Song, A.C. Daly, J.A. Burdick, Jammed microgel inks for 3d printing applications, Adv. Sci. 6 (2019) 1801076, https://doi.org/10.1002/ advs.201801076.
[48] L. Riley, L. Schirmer, T. Segura, Granular hydrogels: emergent properties of jammed hydrogel microparticles and their applications in tissue repair and regeneration, Curr. Opin. Biotechnol. 60 (2019) 1–8, https://doi.org/10.1016/j. copbio.2018.11.001.
[49] M. Hirsch, A. Charlet, E. Amstad, 3D printing of strong and tough double network granular hydrogels, Adv. Funct. Mater. 31 (2021) 2005929, https://doi.org/ 10.1002/adfm.202005929.
[50] M. Kessler, T. Yuan, J.M. Kolinski, E. Amstad, Influence of the degree of swelling on the stiffness and toughness of microgel-reinforced hydrogels, Macromol. Rapid Commun. (2023) e2200864.
[51] M. Kessler, Q. Nassisi, E. Amstad, Does the size of microgels influence the toughness of microgel-reinforced hydrogels? Macromol. Rapid Commun. 43 (2022) 2200196, https://doi.org/10.1002/marc.202200196.
[52] R.L. Gustafson, J.A. Lirio, Binding of divalent metal ions by crosslinked polyacrylic acid, J. Phys. Chem. 72 (1968) 1502–1505, https://doi.org/10.1021/ j100851a018.
[53] T. Zhang, M.S. Silverstein, Doubly-crosslinked, emulsion-templated hydrogels through reversible metal coordination, Polymer 126 (2017) 386–394, https://doi. org/10.1016/j.polymer.2017.07.044.
[54] J. Fan, S.-H. Kim, Z. Chen, S. Zhou, E. Amstad, T. Lin, D.A. Weitz, Creation of faceted polyhedral microgels from compressed emulsions, Small 13 (2017) 1701256, https://doi.org/10.1002/smll.201701256.
[55] T. Heida, J.W. Neubauer, M. Seuss, N. Hauck, J. Thiele, A. Fery, Mechanically defined microgels by droplet microfluidics, Macromol. Chem. Phys. 218 (2017) 1600418, https://doi.org/10.1002/macp.201600418.
[56] J.M. de Rutte, J. Koh, D. Di Carlo, Scalable high-throughput production of modular microgels for in situ assembly of microporous tissue scaffolds, Adv. Funct. Mater. 29 (25) (2019) 1900071.
[57] L.P.B. Guerzoni, J.C. Rose, D.B. Gehlen, A. Jans, T. Haraszti, M. Wessling, A.J. C. Kuehne, L. De Laporte, Cell encapsulation in soft, anisometric poly(ethylene) glycol microgels using a novel radical-free microfluidic system, Small 15 (2019) 1900692, https://doi.org/10.1002/smll.201900692.
[58] V. Chimisso, S. Conti, P. Kong, C. Fodor, W.P. Meier, Metal cation responsive anionic microgels: behaviour towards biologically relevant divalent and trivalent ions, Soft Matter 17 (2021) 715–723, https://doi.org/10.1039/D0SM01458C.
[59] M. Steinacher, E. Amstad, Spray-assisted formation of micrometer-sized emulsions, In Review (2021). https://doi.org/10.21203/rs.3.rs-152490/v1.
[60] A. Sinclair, M.B. O’Kelly, T. Bai, H.-C. Hung, P. Jain, S. Jiang, Self-healing zwitterionic microgels as a versatile platform for malleable cell constructs and injectable therapies, Adv. Mater. 30 (2018) 1803087, https://doi.org/10.1002/ adma.201803087.
[61] B. Kessel, M. Lee, A. Bonato, Y. Tinguely, E. Tosoratti, M. Zenobi-Wong, 3D bioprinting of macroporous materials based on entangled hydrogel microstrands, Adv. Sci. 7 (2020) 2001419, https://doi.org/10.1002/advs.202001419.
[62] H. Yuk, J. Wu, T.L. Sarrafian, X. Mao, C.E. Varela, E.T. Roche, L.G. Griffiths, C. S. Nabzdyk, X. Zhao, Rapid and coagulation-independent haemostatic sealing by a paste inspired by barnacle glue, Nat. Biomed. Eng. (2021) 1–12, https://doi.org/ 10.1038/s41551-021-00769-y.
[63] D. Moon, M.-G. Lee, J.-Y. Sun, K.H. Song, J. Doh, Jammed microgel-based inks for 3d printing of complex structures transformable via ph/temperature variations, Macromol. Rapid Commun. 43 (19) (2022) 2200271.
[64] T.H. Qazi, J. Wu, V.G. Muir, S. Weintraub, S.E. Gullbrand, D. Lee, D. Issadore, J. A. Burdick, Anisotropic rod-shaped particles influence injectable granular hydrogel properties and cell invasion, Adv. Mater. 34 (2022) 2109194, https://doi.org/ 10.1002/adma.202109194.
[65] M.L. Bedell, A.M. Navara, Y. Du, S. Zhang, A.G. Mikos, Polymeric systems for bioprinting, Chem. Rev. 120 (2020) 10744–10792, https://doi.org/10.1021/acs. chemrev.9b00834.
[66] M. Lee, R. Rizzo, F. Surman, M. Zenobi-Wong, Guiding lights: tissue bioprinting using photoactivated materials, Chem. Rev. 120 (2020) 10950–11027, https://doi. org/10.1021/acs.chemrev.0c00077.
[67] Y.S. Zhang, G. Haghiashtiani, T. Hübscher, D.J. Kelly, J.M. Lee, M. Lutolf, M. C. McAlpine, W.Y. Yeong, M. Zenobi-Wong, J. Malda, 3D extrusion bioprinting, Nat. Rev. Methods Primer. 1 (2021) 1–20, https://doi.org/10.1038/s43586-021- 00073-8.
[68] J.-Y. Sun, X. Zhao, W.R.K. Illeperuma, O. Chaudhuri, K.H. Oh, D.J. Mooney, J. J. Vlassak, Z. Suo, Highly stretchable and tough hydrogels, Nature 489 (2012) 133–136, https://doi.org/10.1038/nature11409.
[69] J.P. Gong, Y. Katsuyama, T. Kurokawa, Y. Osada, Double-network hydrogels with extremely high mechanical strength, Adv. Mater. 15 (2003) 1155–1158, https:// doi.org/10.1002/adma.200304907.
[70] W. Li, X. Liu, Z. Deng, Y. Chen, Q. Yu, W. Tang, T.L. Sun, Y.S. Zhang, K. Yue, Tough bonding, on-demand debonding, and facile rebonding between hydrogels and diverse metal surfaces, Adv. Mater. 31 (2019) 1904732, https://doi.org/10.1002/ adma.201904732.
[71] J. Lin, S.Y. Zheng, R. Xiao, J. Yin, Z.L. Wu, Q. Zheng, J. Qian, Constitutive behaviors of tough physical hydrogels with dynamic metal-coordinated bonds, J. Mech. Phys. Solids 139 (2020), 103935, https://doi.org/10.1016/j. jmps.2020.103935.
[72] M. Zhong, Y.-T. Liu, X.-Y. Liu, F.-K. Shi, L.-Q. Zhang, M.-F. Zhu, X.-M. Xie, Dually cross-linked single network poly(acrylic acid) hydrogels with superior mechanical properties and water absorbency, Soft Matter 12 (2016) 5420–5428, https://doi. org/10.1039/C6SM00242K.
[73] S. Anjum, P.M. Gurave, B. Gupta, Calcium ion-induced self-healing pattern of chemically crosslinked poly(acrylic acid) hydrogels, Polym. Int. 67 (2018) 250–257, https://doi.org/10.1002/pi.5517.
[74] L.M. Fuhrer, S. Sun, V. Boyko, M. Kellermeier, H. C¨olfen, Tuning the properties of hydrogels made from poly(acrylic acid) and calcium salts, PCCP 22 (2020) 18631–18638, https://doi.org/10.1039/D0CP02649B.
[75] D.J. Schupp, X. Zhang, S. Sun, H. Co¨lfen, Mineral plastic hydrogels from the crosslinking of polyacrylic acid and alkaline earth or transition metal ions, Chem. Commun. 55 (2019) 4913–4916, https://doi.org/10.1039/C8CC08986H.
[76] A.G. Håti, D.C. Bassett, J.M. Ribe, P. Sikorski, D.A. Weitz, B.T. Stokke, Versatile, cell and chip friendly method to gel alginate in microfluidic devices, Lab Chip 16 (2016) 3718–3727, https://doi.org/10.1039/C6LC00769D.
[77] D.C. Bassett, A.G. Håti, T.B. Melø, B.T. Stokke, P. Sikorski, Competitive ligand exchange of crosslinking ions for ionotropic hydrogel formation, J. Mater. Chem. B 4 (2016) 6175–6182, https://doi.org/10.1039/C6TB01812B.
[78] J.P. Gong, Why are double network hydrogels so tough? Soft Matter 6 (2010) 2583–2590, https://doi.org/10.1039/B924290B.
[79] J.P. Gong, Materials both tough and soft, Science 344 (2014) 161–162, https://doi. org/10.1126/science.1252389.
[80] G.E. Fantner, T. Hassenkam, J.H. Kindt, J.C. Weaver, H. Birkedal, L. Pechenik, J. A. Cutroni, G.A.G. Cidade, G.D. Stucky, D.E. Morse, P.K. Hansma, Sacrificial bonds and hidden length dissipate energy as mineralized fibrils separate during bone fracture, Nat. Mater. 4 (2005) 612–616, https://doi.org/10.1038/nmat1428.