¶ MEA-seqX:大规模电生理和转录网络动态的高分辨率分析
Brett Addison Emery, Xin Hu, Diana Klütsch, Shahrukh Khanzada, Ludvig Larsson,Ionut Dumitru, Jonas Frisén, Joakim Lundeberg, Gerd Kempermann, and Hayder Amin*
B. A. 埃默里、X. 胡、D. 克吕奇、S. 坎扎达、H. 阿明
德国神经退行性疾病中心 (DZNE)
“生物混合神经电子学” 课题组
地址:塔茨贝格街 41 号,01307 德累斯顿,德国
电子邮件:hayder.amin@dzne.de
L. 拉尔森、J. 隆德伯格
生命科学实验室
基因技术系
瑞典皇家理工学院
地址:汤姆特博达大道 23 号,17165 斯德哥尔摩,瑞典
I. 杜米特鲁、J. 弗里森
细胞与分子生物学系
卡罗林斯卡学院
地址:贝采利乌斯路 35 号,17165 斯德哥尔摩,瑞典
G. 肯珀曼
德国神经退行性疾病中心 (DZNE)
“成体神经发生” 课题组
地址:塔茨贝格街 41 号,01307 德累斯顿,德国
德累斯顿工业大学再生治疗中心 (CRTD)
地址:费彻尔街 105 号,01307 德累斯顿,德国
H. 阿明
德累斯顿工业大学
卡尔·古斯塔夫·卡鲁斯医学院
地址:山街 53 号,01069 德累斯顿,德国
本文作者 ORCID 识别码可在 https://doi.org/10.1002/advs.202412373 下找到。
2025 作者。Advanced Science 由 Wiley-VCH GmbH 出版。这是一篇基于知识共享署名许可条款发布的开放获取文章,允许在任何媒介中不受限制地使用、分发和复制,但必须正确引用原文。
脑功能的概念意味着分子事件与神经元活动之间的相互一致性和因果影响。从并发的分子和电生理网络事件中解码纠缠信息,需要跨越尺度和模态的创新方法。MEA-seqX平台整合了高密度微电极阵列、空间转录组学、光学成像和先进计算策略,能够在介观空间分辨率下同步记录分析分子与电生理网络活动。将该平台应用于经验依赖性可塑性的小鼠海马体模型时,MEA-seqX揭示了转录与功能间极大增强的嵌套动力学。图论分析显示密集连接的双模态枢纽节点增多,这标志着首次在分子和功能水平同步观测到海马环路的协调动力学。该平台还能根据不同的双模态特征识别细胞类型。通过机器学习算法,仅凭空间基因表达数据即可精准预测全网络电生理活动特征,实现了以往难以达成的跨模态、跨时间、跨尺度的融合分析。
¶ 1. 引言
大脑进化以稳健且高效地处理复杂信息,从而维持体内平衡、适应环境、做出决策并执行高级认知功能。[1] 要系统性地理解大脑从分子到系统层面的复杂性,必须整合来自不同时空背景的多模态数据。[1] 这一宏伟目标的核心在于实现神经元电生理与分子表型的网络级和细胞级整合,因其既是生理功能的基础,也与神经发育及神经退行性疾病密切相关。[2,3] 为此,诸如补丁测序(Patch-seq)等方法学进展,已实现单细胞转录组测定,并在电生理记录后完成单个神经元的形态重建。[4,5] 尽管意义重大,但Patch-seq受限于其极低的通量以及无法解析更大空间尺度上的神经网络。同样,将柔性生物电子学与原位RNA测序相结合以绘制电活动和基因表达图谱的Electro-seq技术,仅限于体外神经元培养,缺乏在复杂组织环境中的适用性。[6] 另一现有方法是CaRMA,该方法结合钙成像与RNA-FISH技术来绘制基因表达与神经活动图谱[7]。然而与其他方法类似,CaRMA受限于钙成像缓慢的时间分辨率以及事后转录组分析,这制约了分子数据与功能数据的实时整合。其应用也局限于小型脑区域,限制了其对更大更复杂网络的可扩展性。
然而,两项独立研究为这一问题提供了新视角。高通量空间分辨转录组学(SRT)通过在保留空间组织架构的同时分析数千个基因的表达模式,为关注脑区的分子多样性提供了前所未有的洞察力[8,9]。但SRT技术的时间分辨率有限,仅能提供转录组景观的静态快照。时空转录组学必须通过不同时间点的多个样本才能解析[10,11],这意味着分子特征相关的功能状态需要通过间接证据进行推断。这种方法显然无法充分解析电信号与分子信号的多样性及其在脑功能、认知和行为背后的相互作用机制[12,13]。 但基于高密度互补金属氧化物半导体CMOS)生物传感微电极阵列(CMOS-MEA)的新型脑芯片技术,现已能够实现无创、多位点、长期、无标记的同步检测:在保持细胞完整性的前提下,以高时空分辨率同步捕获数千个神经元的局部场电位和脉冲活动[14–19]。整合这些尖端技术将强化对同一组织样本的解码能力,在保留细胞空间定位的同时,协同解析时空电生理信息与空间转录组数据。其基础科学假说在于:脑功能的实现依赖于大型神经元集群与基因网络的协同作用,这些网络遵循共同的组织原则[20],在跨尺度的时空维度上进行信息处理[13,21],并随着经验积累与疾病状态发生动态演化。
为验证此预测,我们开发了MEA-seqX平台。该平台结合脑芯片记录与空间测序技术,在跨尺度计算框架中利用光学成像进行空间参照对齐。MEA-seqX能够依次实现以下功能:以高时空分辨率获取急性脑切片中大细胞集群同步放电模式的电生理记录;对完整神经环路进行空间定位成像;对同一神经环路进行多重细胞转录组分析。这揭示了转录动态表现为基因表达在不同网络状态、条件及时间推断分子轨迹中的结构化变异,从而建立转录组状态与神经活动之间的功能关联。通过采用自动机器学习算法,并在高分辨率表征中保留时间与拓扑信息,MEA-seqX能够实现:指定空间转录组网络与连接性及其他多模态数据的实时关联;基于底层放电信息量化伪时间衍生的时空分子动力学;解卷积空间分辨的细胞类型组成;以高精度根据转录组谱预测电生理网络活动特征。MEA-seqX平台通过实时提供分子与功能数据的高分辨率同步记录,突破了现有技术的局限。这使研究人员能够对大网络中快速与缓慢的神经过程进行全面分析,为基因表达与神经元活动的协调机制提供新见解。该平台整合多尺度数据的能力,使其成为推动基础研究和临床应用(包括生物标志物发现和精准医疗)的强大工具。
为了阐明这种方法的力量,我们将其应用于经验依赖性可塑性的经典丰富环境范式——该范式中唯一的实验干预在于实验室小鼠差异化饲养条件。与标准饲养的小鼠相比,丰富环境组小鼠生活在由同源基因动物组成的大型群体中,活动空间更为广阔[17,22]。这种简洁却极具影响力的范式能引发全脑及海马体的结构与功能改变,深刻影响着科学界与公众的认知。我们近期研究发现,其在出人意料的尺度上产生的影响与(海马神经环路层面的改变相关[17]。如今MEA-seqX技术使我们得以探索一个曾经无法触及的问题:大规模海马网络的计算动力学与连接组如何与底层转录动力学相互关联。我们的假设认为这两种动力学之间存在因果联系——这种关联在生物医学语境中常被提及,但以往仅靠零星有限的数据点支撑。MEA-seqX通过提供更坚实的基础改变了这一现状。
我们的研究揭示了识别大规模神经环路分子特性与动态变化的潜力。我们期待在健康与疾病背景下开发出具有高功能有效性的新型多模态模型。这类"生物标志物"为开发新型诊断与筛查工具带来巨大潜力,尤其在(但不限于)精准医疗领域。
¶ 2. 结果与讨论
¶ 2.1. 接口技术与信息整合——从转录组到功能网络
MEA-seqX通过在高密度CMOS微电极阵列(CMOS-MEA)[14,17,23]上实现网络电生理学(n-Ephys),整合了脑芯片技术;同时通过空间分辨转录组学(SRT)融合空间测序技术,并借助光学显微镜与生物成像进行联合定位(图1a-d及附图S1a,支持信息)。该系统从 厚的小鼠海马-皮层“HC”急性切片中获取了细胞外放电模式的高分辨率时空记录,这些切片与4096个片上传感器相连接。同步采用光学成像实现精确解剖定位,并获取了神经环路内同一细胞的高分辨率转录组图谱数据。
对我们转录组数据集的质量控制显示,在使用均匀流形逼近与投影(UMAP)方法进行可视化时,具有相似的nFeature与nCount RNA统计特征及组织结构(图S2a-d,支持信息)。[24] 通过主成分分析(PCA)和K均值聚类算法评估了海马体全网络活动,这些方法揭示了各相互连接海马体层中振荡波形及其形态的独有特征(图S2e,支持信息)。[22] 通过基于Python的计算流程对多维读数进行处理,以高空间分辨率定量绘制电路的分子动力学图谱(图S1b,支持信息)。我们开发了多尺度空间重标定与配准流程,以在n-Ephys电极-SRT位点界面与其各自全网络范围的功能性电活动及转录组特征读数之间建立直接对应关系,并确定其在组织中的定位。该过程通过自动缩放算法实现,采用基于光学成像的图像尺寸调整与旋转技术,结合n-Ephys电极-SRT位点界面的物理尺寸以及齿状回中的两个解剖学标志(图1e;详见实验方法部分)。最终生成的叠加图谱使得我们能够从转录组读数中识别网络特征,并与神经活动读数实现对齐(图1g)。MEA-seqX生成了多模态数据连接的精确拓扑图,呈现了SRT位点与底层放电电极的局部和全局关系(图1g)。基于海量高维数据,我们推导出放电信息流背后的转录伪时序动态(图1g)。通过整合反卷积方法,从空间转录组数据中推断出细胞类型分辨率,并将神经元异质性与放电特征进行关联(图1g)。最终,MEA-seqX提供自动机器学习算法,可通过空间转录组图谱预测高精度网络电生理活动特征(图1g)。这种电生理记录与转录组数据的整合为全网络范围的分子和功能活动研究提供了前所未有的深度洞察。值得注意的是,独立的数据采集方法(例如为每种模态使用不同组织切片)将无法达到最佳效果,并且在捕捉神经元功能与基因表达之间精确空间解析的相互作用方面存在根本性局限。若不能保留转录程序展开所依赖的精确细胞与网络拓扑结构,推断出的关系将仍是推测性的,而非直接可测量的。因此,在同一组织样本中同步获取电生理学和转录组学数据,可确保观测到的功能-转录关系反映真实的相互作用,而非源于不同样本的变异。基于这种整合方法,后续章节通过将空间转录组与功能动力学相关联,拓展了这些发现,揭示了海马体中转录网络如何与电生理特征相吻合。
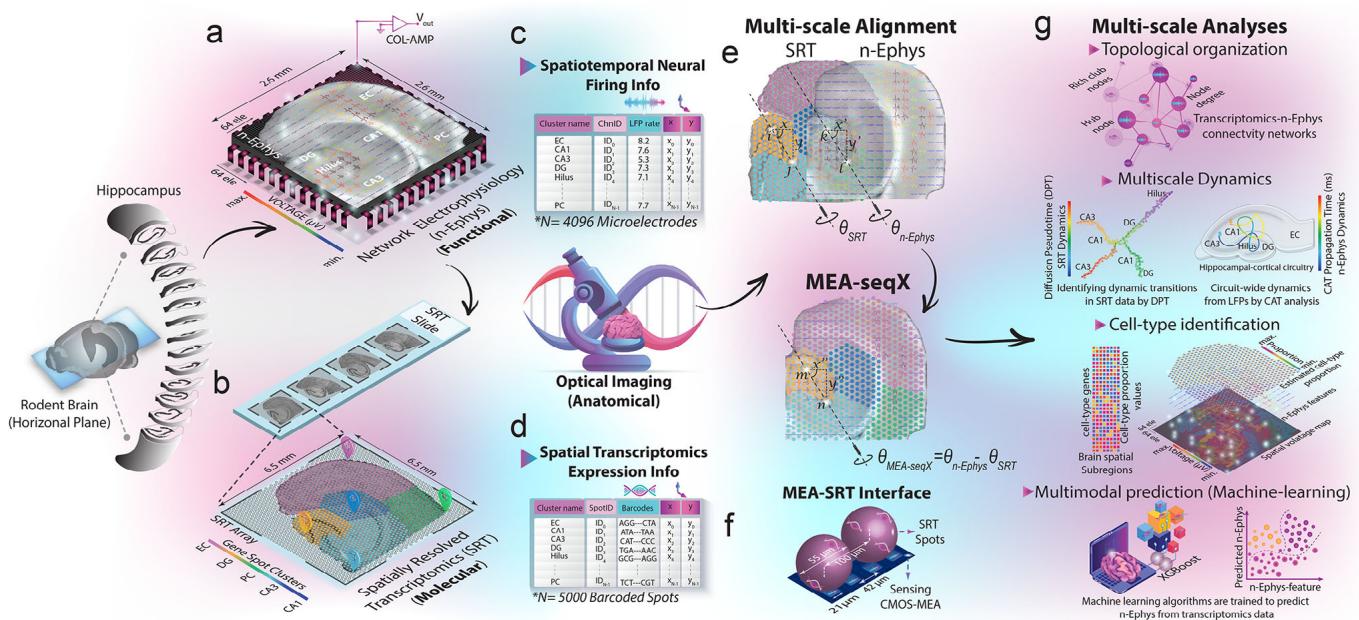
图1. MEA-seqX平台整合跨尺度脑动力学概览。a,b) 该平台结合高密度CMOS微电极阵列、光学显微镜与空间测序技术,旨在解析空间转录组与神经振荡动力学之间的关联。c) 配备4096个传感电极的高密度CMOS微电极阵列能以亚毫米级空间精度,精准捕获源自海马体各区域的功能性放电LFP模式。d) 基于空间条形码技术的SRT平台包含5000个检测点,可实现基因表达谱的系统性空间映射,揭示海马体各区域的转录组空间异质性。e) 采用Python开发的数据处理流程通过多层面空间重标定与配准方法,构建神经活动与转录组特征间的直接关联。f) 以单个传感电极与SRT检测点为例的局部特写,凸显了两类平台在物理尺度上的显著差异。g) 基于多维读数的高级分析可揭示多尺度网络特征:拓扑图分析捕捉多模态转录-功能连接网络,基因伪时序与活动轨迹中心量化多尺度神经动力学,空间转录组数据推断的细胞类型组成与其在电生理记录中获取的放电特征相关联,自动化机器学习算法能根据转录组谱精准预测电生理特征。
¶ 2.2. 空间转录组与全网络神经功能动态的关联
为了将这一流程应用于一个具体的、先前无法解决的研究问题,我们采用MEA-seqX框架来揭示经验依赖性可塑性[17]对神经元集群协同活动及其与协调转录活动相互作用的影响。我们分别从标准环境(SD)和富集环境(ENR)饲养的小鼠中制备海马切片,用于记录局部场电位(LFP)的振荡模式、光学成像及空间相关性转录(SRT)测序。为评估基因表达的空间模式(SRT)如何与同一脑组织的功能性网络电生理活动特征(n-Ephys)相对应,我们通过测量斯皮尔曼相关系数[25]来量化SRT位点基因表达谱的转录相似性,并检验其与功能性网络活动特征(即振幅、LFP事件延迟、能量、LFP频率、负峰计数和正峰计数)的关联。研究发现,与SD组相比,ENR组中与功能耦合的齿状回(DG)和CA3海马亚区对应的基因表达模式显著增强(图2a)。特别是在ENR组中,DG、CA3和CA1亚区内与LFP频率相关的转录本数量分别较SD组增加了2.2倍、2.5倍和0.6倍(图2b)。我们进一步探究哪些特定基因会驱动分子网络与功能网络之间更强的因果联系。基于基因本体聚类,我们将相关基因归类为六个靶向基因家族,包括即刻早期基因(IEGs)、海马神经发生、海马信号通路、受体与通道、突触可塑性、突触小泡和粘附[26,27]。对这些家族转录本的分析表明,与SD组相比,ENR组的IEGs表达、离子通道活性、突触功能和神经发生均有增强。对海马活动和功能至关重要的基因(如Bdnf、Egr1、Homer1, Npas4, Gria2及Campk2a)在ENR组海马转录组中的表达水平均高于SD组(支持信息图S3)。
接下来,为识别整合性SRT与n-Ephys模态中不同的时空模式,我们采用稀疏约束非负矩阵分解(NMF)[28]的无监督机器学习算法。该方法能够将多模态数据分解为差异表达的基因子网络、空间位点及网络电生理特征集合,从而实现降维与结果可解释性(图2c)。即刻早期基因(IEGs)在连接空间转录模式与局部场电位活动方面表现出显著贡献(图S3,支持信息)。为确定NMF分解中降维模型需识别的最优组件数量(即因子p),我们评估了在不同模型复杂度下重建V矩阵的效果。重建误差[29]的两个线性区间在 处呈现明显拐点,此时增加更多模式仅能边际提升拟合质量(图2d)。输入的V表达矩阵包含SRT与n-Ephys(即LFP频率)数据的整合信息,每个条目代表特定空间位点中与网络功能特征相关联的基因表达水平(图2e),该矩阵被分解为两个非负矩阵:基底W矩阵包含分解后的空间基因表达模式及其位点信息,反映功能特征在空间位点间的贡献分布(图2f);系数H矩阵则表征基因表达模式对每个空间位点及n-Ephys功能特征谱的推断贡献度(图2g)。本分析揭示了ENR网络中标准化基因表达与网络特征值的增强,以及驱动基因表达强度的空间解析组件提升。此外,我们还发现空在SD和ENR网络中部分特定子网络的研究中,凸显出具有主导贡献的基因,例如Bdnf、Egr1、Fosb和Npas4。
MEA-seqX表明,经验依赖性动力学在神经元群体活动与其相应转录模式的协调相互作用中具有计算功能。在确立了空间基因表达与功能网络活动之间的强相关性后,下一节将探讨这些相互作用在海马网络内的组织方式。我们通过研究多模态网络的拓扑结构,揭示了分子子网络与电生理子网络之间的协调组织关系。
¶ 2.3. 空间分辨转录组和活动模式的协调拓扑网络组织
我们采用定量测量方法,全面考察了在睡眠剥夺(SD)和环境富集(ENR)条件下,从空间转录组(SRT)数据获得的海马体子网络与神经电生理(n-Ephys)记录之间的相互联系。通过计算“互信息”[30](即两个变量间相互依赖程度的度量),我们评估了特定基因家族内基因表达的相互依赖程度(图S4a,支持信息);同时采用皮尔逊相关系数(PCC)[14,17]来衡量成对放电电极间的互协方差关系(图S4b,支持信息)。这些计算使我们能够建立多尺度的连接矩阵。随后,我们计算了每个位点中目标基因家族的互信息距离分数,以评估不同位点间相互作用模式的差异。将这些分数进行跨位点比较后,我们将其归类为不同簇群(图S5a-f,支持信息)[30]。与此同时,通过分析互连海马体层中同步局部场电位(LFP)活动的皮尔逊相关系数,我们对相关矩阵的差异进行了量化(图S5g,支持信息)[14,17]。
在分析多种基因家族时发现,与SD相比,ENR转录组在单个海马亚区内部以及不同海马亚区之间均表现出更高的互信息。这表明海马子网络内基因表达模式之间的协同活动与通信具有更稳健的统计关联(图S5a-f,支持信息)。重要的是,从LFP互相关图[17] ((图S5g,支持信息))可以明显看出,这一发现与ENR网络相较于SD网络中局部和全局时空相互作用强度的显著增强高度吻合[17]。 海马网络内的局部与全局互连性反映了转录组表达与神经活动之间的协同动力学。局部层面,这种协调性体现在DG、CA1、CA3等特定亚区内部,其中转录模式与LFP活动在这些小尺度网络中紧密关联。全局层面,这些亚区之间的长程协调性增强,揭示出一个横跨多个海马-皮层区域的更紧密互连的网络整体。由ENR中经验依赖性可塑性驱动的这种增强协调,最终形成了海马内部更稳固的功能与分子层面通信。这些发现表明ENR独立地增强转录和电生理网络一致性,从而在分子和功能层面产生平行的、功能上一致的适应性变化。
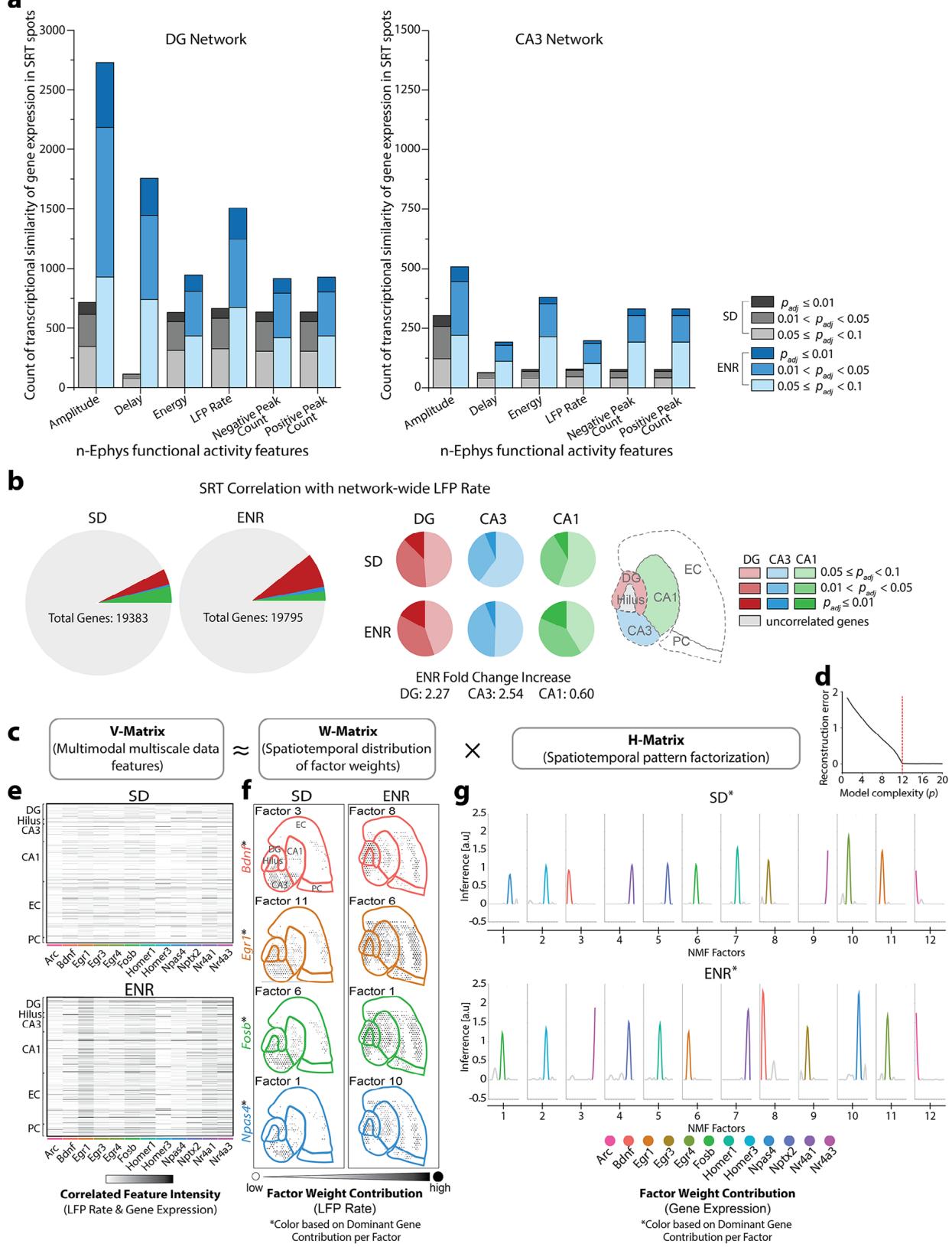
图2. 空间基因表达模式与网络电生理特征的整合。a) SRT表达模式与功能性n-Ephys活动特征的比较分析显示,与SD相比,ENR中对应于DG和CA3区域的基因表达模式增强。SRT和n-Ephys特征相关性的统计显著性使用Benjamini-Hochberg错误发现率(FDR)调整的p值 )进行量化。b) 基于全网络LFP速率的相关基因定量分析显示,与SD相比,ENR中DG区域的相关基因增加了2.2倍,CA3和CA1亚区域分别增加了2.5倍和0.6倍。SRT和LFP相关性的显著性使用
随后,为描述和量化多模态转录-功能读数中的拓扑组织、信息流与通信特性,MEA-seqX采用图论方法评估多尺度网络拓扑指标(图3a)。该方法通过构建详细的接线图,描绘转录组学与神经活动模式之间的局部和全局互连关系。其中转录组图谱基于互信息评分构建,功能图谱则源自睡眠剥夺(SD)和环境富集(ENR)条件下局部场电位所捕获的神经元集群共激活连接模式。由此产生的转录组-神经功能连接图谱,揭示了源自空间即刻早期基因(IEG)与时空功能神经活动的互连子网络空间排布。值得注意的是,这些图谱在不同尺度和模式下均展现出相似的空间分布与连接模式(图3b,c)。通过对比SD与ENR条件下的海马体网络,进一步凸显出跨尺度因果协调水平的提升。相较于SD,ENR条件下的转录-功能连接组呈现出增强态势。
我们还对构建的图谱进行了分析,以识别高度互连的节点(称为"枢纽复合体")及转录功能连接组内被称为"富人俱乐部组织"的密集连接枢纽[31]。与标准饲养组相比(图3d,e;gray),源自空间转录组和神经元电生理数据的ENR子网络展现出更强的枢纽复合体互连性和富人俱乐部组织性(图3d,e;blue)。这表明丰富环境体验能增强协调互动的专业化程度,提升网络韧性,并扩大跨转录功能尺度的全局通信能力[32]。这种专业化表现为模块化程度提升、枢纽结构显著形成及富人俱乐部连接增强,共同反映出ENR条件下海马网络具有更明显的层级化组织和功能优化特征。这些发现为分子与功能枢纽复合体之间的动态相互作用提供了新见解,对理解多尺度拓扑网络组织的整体协调机制具有重要贡献。
许多生物网络具有小世界拓扑结构[33],其特点在于采用无标度架构——该架构由高度连接的枢纽节点构成,且度分布呈现幂律尾衰减特征[34]。通过分析SD与ENR网络中转录组学与功能连接组的互连链路累积度分布,我们发现这些多尺度分布确实遵循幂律函数,我们先前在成年海马神经发生的转录组网络[35]和ENR中的全网络活动[17]中也曾提出过这一假设。与SD相比,ENR网络中SRT和n-Ephys分布均表现出更重的尾部,表明存在更多密集连接的枢纽节点(图3f及插图)。该发现得到了多尺度度分布的稀疏约束非负矩阵分解分析的支持(即类似于图2c-g所采用的方法)。我们通过量化多尺度分解的基因可变表达子网络、空间位置集合及网络连接节点度特征,识别出与空间分辨的即刻早期基因表达及网络连接程度相关的拓扑指标变异(图S6,支持信息)。
通过在一次实验中揭示经验诱导的海马体跨尺度连接组及其复杂的多层次动态特征,我们的研究结果证明了MEA-seqX技术具备整合捕获协调转录组与功能数据的能力。这种整合为神经通信、韧性水平、层级结构和多尺度特异性提供了新见解,而这些领域以往只能基于有限数据进行独立研究。[32,36,37]
在丰富环境下海马体网络增强的拓扑结构凸显了转录动态与功能动态之间多尺度协调的力量。下一节中,我们将评估这些相互作用的时间动态,重点关注分子和功能信息如何随时间展开以驱动协调的神经活动。
¶ 2.4. 多模态信息的多尺度动态特性评估
为揭示跨尺度和跨模态的同步动力学这一挑战,我们融合了两种前沿计算技术——面向空间转录组学的扩散伪时间分析[38]与面向多维电生理学的活动轨迹中心重构[14,17,39,40]。该整合方法旨在解析海马环路中基因表达的时间进程及全网络神经活动模式。我们通过对静态快照式空间转录组数据实施扩散伪时间分析,依据表达相似性重构位点分布从而实现伪时间排序,由此构建了空间转录组发育轨迹的网络表征。通过计算扩散映射空间中基于向量的随机距离的欧几里得距离,我们推算出分化概率,这有助于从高维观测数据中识别低维动态变化。我们在此重点研究了IEG在SD和ENR区域的表达情况,结果揭示了基于IEG表达的DPT存在显著的区域差异(图4a、b)。在SD和ENR中,基于IEG表达的DPT显示出显著的区域差异(图4a,b)。同时,我们通过构建全网络n-Ephys振荡活动的CATs,量化了海马内的时空传播路径,并由此计算出这些放电模式的时空位移速率(图4c,d)。CAT持续时间分析表明,与先前报道一致,ENR中放电事件的传播速度较SD更快(即持续时间更短)[17]。值得注意的是,SD与ENR的转转录组显示,与睡眠剥夺组相比,丰富环境转录组在所有四个海马区均表现出更快的伪时间轨迹,这反映了在丰富环境组神经电生理连续积累测试中观察到的更快时空传播模式(图4c–e)。[17]通过将伪时间轨迹空间图谱与神经活动轨迹的时间进程相整合,并考量丰富体验的调节作用,我们得以从多尺度视角阐释分子过程与电生理过程如何在生物系统中同步展开并相互作用。深入探究经验在依赖活动动态的情况下,SD与ENR之间的对比尤其具有启发性,因为这些差异化参与模式凸显了反映潜在功能和分子适应的不同子区域招募。这样的比较有可能揭示分子水平动态变化与功能尺度动态变化之间的因果联系。
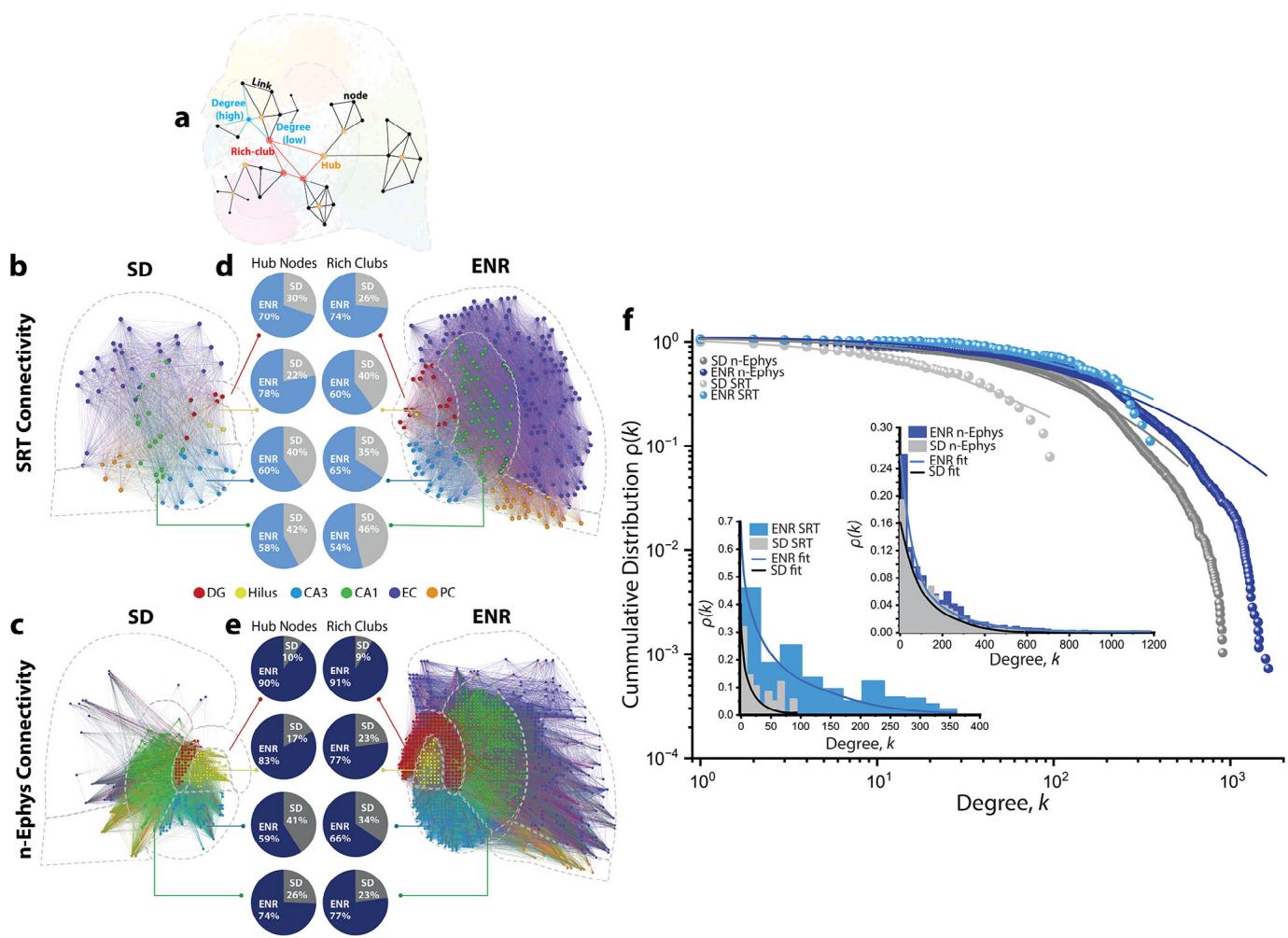
图3. 空间测序连接组与神经功能连接的多尺度网络拓扑分析。a) 关键多尺度图度量示意图,展示了基于急性海马-皮层切片中互联程度定义的特征性枢纽节点和富人俱乐部节点。节点度对应特定节点上的连接链路数量。b) ENR和SD条件下海马互连层中空间即刻早期基因的连接图谱。使用Gephi可视化网络,显示完整测序SD数据(节点 ,连接 )和ENR数据(节点 ,连接 )的总连接。c) ENR和SD条件下海马互连层中时空功能神经活动的连接图谱。使用Gephi可视化网络,显示SD总连接的 (节点 ,连接 )和ENR大规模记录数据(节点 ,连接 )。(b)和©中的图节点按度强度缩放,并根据海马模块关联性着色(见彩色图例)。彩色连线标识簇内与簇间连接。d) SD和ENR不同海马转录组网络中量化枢纽节点与富人俱乐部节点的百分比。e) SD和ENR不同海马功能网络中量化枢纽节点与富人俱乐部节点的百分比。f) 幂律分布表明SD和ENR网络中具有小世界特征的无标度转录组(SRT)-功能(n-Ephys)网络拓扑。ENR网络(SRT与n-Ephys;blue)累积连接分布的双对数图显示出比SD网络(SRT与n-Ephys;gray)更显著的厚尾特征,表明低度节点与少数高度连接枢纽共存,且程度高于达到截断值的SD网络( ,Kolmogorov-Smirnov检验)。线性尺度插图也支持这一结论,其幂律符合性通过帕累托拟合评估( ,Kolmogorov-Smirnov检验)。对数正态函数在双对数图中拟合幂律分布的优度[决定系数 分别为SRT、n-Ephys(SD)、SRT和n-Ephys(ENR)的0.95、0.98、0.96和0.97]。帕累托概率密度函数在线性图中拟合幂律分布的优度 分别为SRT、n-Ephys(SD)、SRT和n-Ephys(ENR)的0.96、0.97、0.95和0.97]。
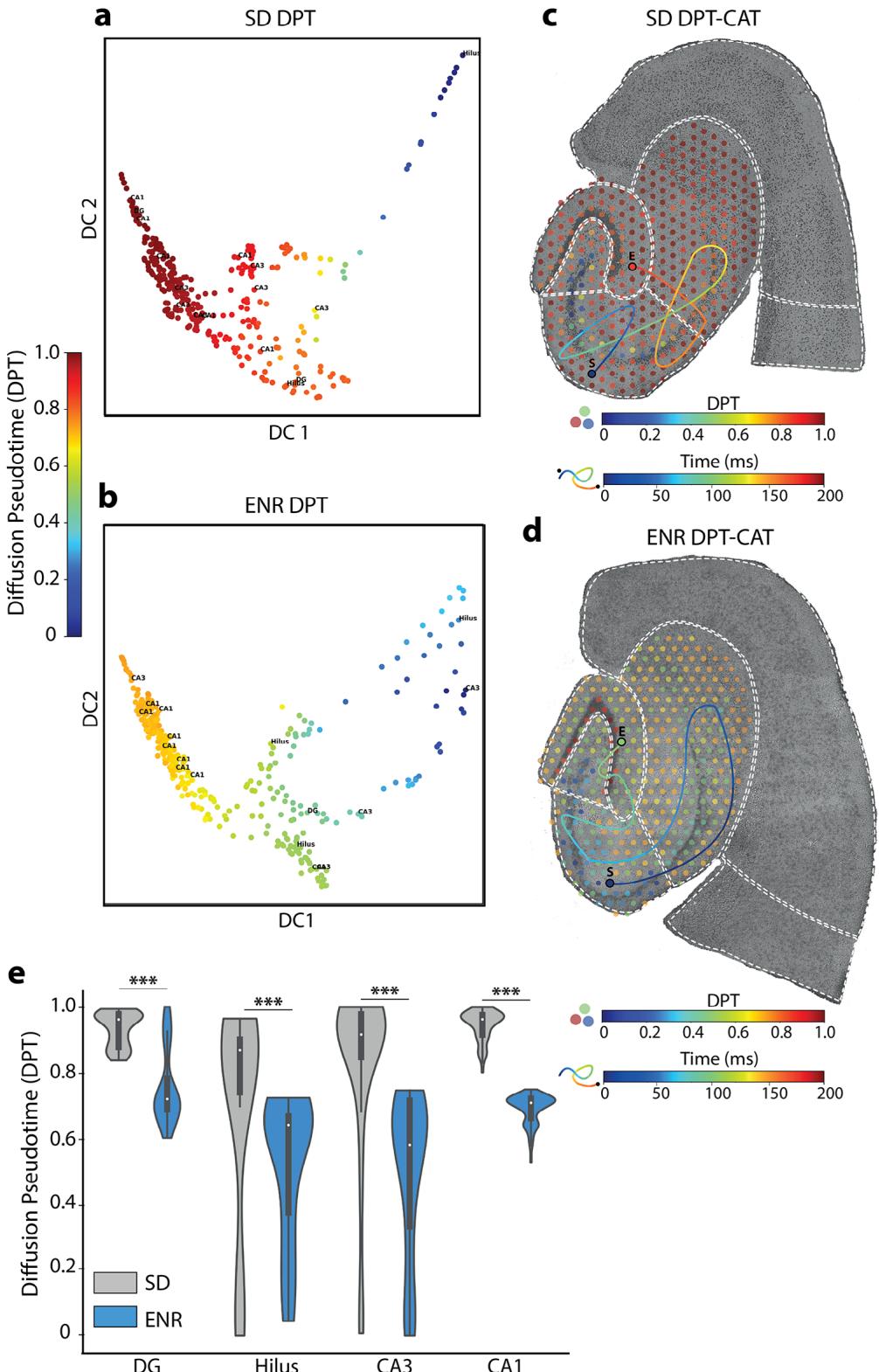
图4. 海马体多尺度动力学分析。a) 基于SRT SD数据的即刻早期基因表达中DPT的差异进展,展示了海马空间区域内细胞的发育轨迹。b) 与(a)相同但基于ENR数据。c) 通过SD数据中DPT与CAT分析的对应关系推断共定位、时空对齐及功能洞察。d) 与©相同但基于ENR数据。e) 量化SD和ENR中互联海马区域的DPT差异进展( ,ANOVA检验)。
以SD和ENR为代表的不同体验环境,在转录组层面诱发差异化响应,这种响应与同一细胞中的神经活动相互关联、不可分割。这些发现直接证明了转录组与功能性神经元活动之间多尺度协调的差异化运作,同时保留了不同环境(SD与ENR)的空间信息。如更快的DPT和神经元放电时空位移(CAT)的减少所示,分子过程与功能过程之间的这种对齐在ENR条件下显著增强。我们的研究揭示了环境富集如何促进基因表达与神经元活动之间更同步的协调,从而加强网络整合与可塑性。这些结果基于先前关于经验如何调节突触可塑性与内在可塑性的研究[41],并通过揭示基因表达如何直接影响网络同步与功能动力学拓展了这一认知[27,42]。此外,我们的研究支持多尺度协调在记忆痕迹形成与认知韧性中的作用[43]。
随着分子和功能活动的时间动态图谱绘制完成,下一部分将深入探究海马回路中的细胞多样性。通过识别特定细胞类型并将其与功能和转录组图谱相关联,我们得以更深入地理解驱动海马网络动态的细胞异质性。
¶ 2.5. 时空细胞类型识别
接下来,为理解神经细胞类型的转录多样性及其在海马回路放电模式中的作用,[44]我们采用基于条件自回归的反卷积方法(CARD),以单细胞测序数据作为参考。[45,46]通过反卷积获得的基因表达模式,我们得以确定细胞类型及局部组织构成,进而构建多尺度空间图谱,揭示同一海马组织中神经类型的异质性及其放电特征。CARD的初步应用实现了海马细胞类型的广泛类别划分,该多样化群体包含星形胶质细胞、内皮细胞、室管膜细胞、巨噬细胞、小胶质细胞、神经源性细胞、神经元、少突胶质细胞以及NG2细胞。在未进行任何筛选前,我们识别出85种不同细胞类型,这一发现印证了切片获取技术的实验有效性。经过低计数细胞类型的剔除后,我们最终获得由76种细胞类型组成的稳健集合。
有趣的是,当这些细胞类型暴露于来自SD和ENR的两种不同转录组输入时,我们观察到两种转录组中主要细胞类型的分布具有一致性(支持信息图S7)。这一结果凸显了我们方法的稳健性以及研究结果的可重复性。此外,通过整合CARD方法MEA-seqX技术对于实现空间分辨的细胞类型组成并将其与大规模振荡电生理特征相关联发挥了关键作用(图5a)。我们根据独特的标记基因,在齿状回(DG)和CA3区鉴定了特定高比例细胞类型。在DG中,颗粒细胞(GCs)以Cck和Penk表达为特征,而CA3锥体细胞则呈现Nos1和Inhba标记。与既往研究一致[47],我们观察到DG分子层和上锥体层中同时存在常见的Cck表达GCs与较少见的Penk表达GCs(图5b)。值得注意的是,这两种标记基因在ENR条件下的表达量均显著高于SD组(图5d)。当将这些标记基因叠加至DG功能网络数据时,ENR样本显示出增强的放电模式和信号幅度,尤其在DG上锥体层内更为明显(图5b,d)。已有研究将Penk与DG印迹细胞富集及其在海马相关行为中的参与相关联[47],而Cck is则涉及DG中细胞集群的动态选择与控制[48]。我们的数据与现有报道相符,并突显了ENR条件下DG兴奋性和时间动态的显著提升,这为探索丰富经验如何影响海马环路中转录-功能互作提供了潜在途径,有助于理解记忆的细胞分子基础[49]。 通过对CA3锥体细胞层空间分布进行类似分析(图5c),我们鉴定出Nos1和Inhba标记基因在ENR中的表达显著高于SD(图5e)。这种转录组读数与CA3区内增强的局部场电位频率及独特波形特征相吻合(图5c,e)。Nos1已被证实与关键神经机制相关,包括长时程增强(LTP)、突触可塑性及神经环路动态调控[50,51],而Inhba则与神经保护和神经元存活相关[52]。这些报告为我们在ENR组经验依赖范式框架内获得的增强型转录-功能发现提供了有力佐证。这进而为更深入探索这些标记基因在CA3锥体神经元中的具体作用及其对跨尺度神经功能与失调理解的潜在意义铺平道路。 超越单细胞研究方法并聚焦多尺网络水平动态,我们的发现提供了对海马细胞类型及其跨尺度交互序列电生理特性更全面而细致的理解。
最后,我们在下一节中利用机器学习模型来预测基因表达谱如何影响网络范围内的电生理特征。这种方法提供了一个强大的工具,以进一步阐明分子和功能尺度之间的因果关系。
¶ 2.6. 跨尺度和模态的预测
为了探究单个空间分辨基因的表达谱是否能预测海马网络范围内的电生理活动特征,我们采用了梯度提升(XGBoost)算法,该算法以其强大的互通过整合多个树模型,我们提升了模型的可解释性。[53] 先前的研究方法主要利用低通量转录组学与电生理技术(如单细胞RNA测序和膜片钳),聚焦于与细胞类型间电生理及形态多样性相关的基因属性;[4,5,44] 而本研究旨在评估是否可利用空间分辨转录组数据预测特定功能网络活动特征。我们使用检测到的SRT斑点中 的空间转录组数据点(即HC组织空间背景下的333个基因及六个基因家族作为输入),针对每个定量n-Ephys特征训练XGBoost模型。基于差异空间基因表达,成功预测了三个时空n-Ephys活动特征(LFP频率、振幅和LFP事件延迟)。XGBoost模型一经训练即可自动运行,这意味着在数据保持一致的条件下,无需额外人工输入即可根据空间分辨基因表达数据预测全网络电生理活动特征。但引入新基因列表或n-Ephys活动特征需重新训练模型(详见实验方法部分),以准确捕捉新的预测关系。这种自动化机制能高效处理新数据集,确保在多实验条件下具有可扩展性且性能稳定。
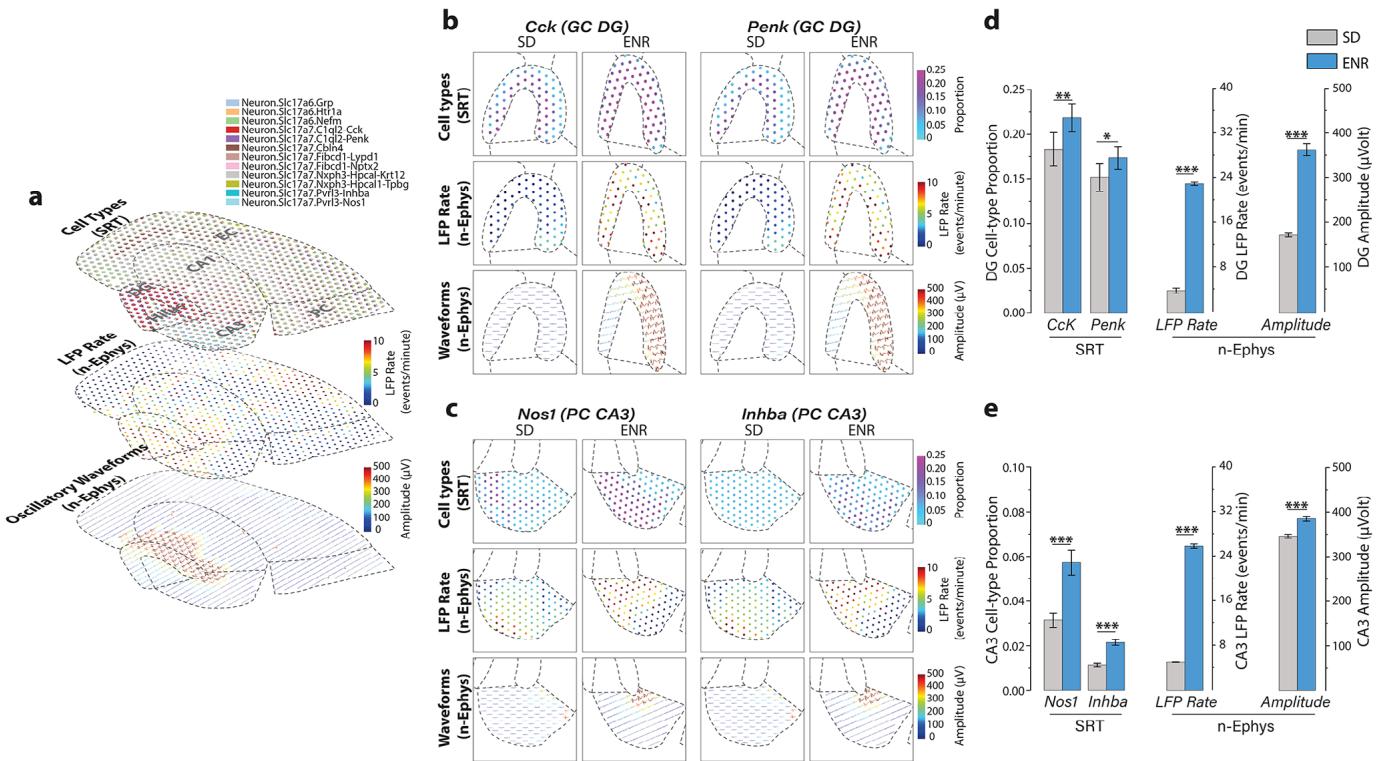
图5. 空间细胞类型及其放电模式特征分析。a) 将CARD方法整合至MEA-seqX平台可获得具有空间分辨率的细胞类型组成及其振荡放电特性。该展示包含通过SRT读数、LFP频率以及从全海马网络n-Ephys读数得出的振荡波形所确定的占比前20位的细胞类型。b) 针对齿状回区域的检测提供了细胞类型空间分辨率组成及其相应振荡放电特征的深度视图。c) 同样地,在CA3锥体细胞网络内进行的区域评估揭示了细胞类型空间组成及其相应振荡放电特征。d) ENR转录组在齿状回颗粒细胞类型中显示出更高比例的空间定位标记基因Cck和Penk,相较于SD转录组显著升高。同时ENR表现出比SD更高的LFP频率与振幅 , , ANOVA, , ANOVA)。e) CA3区域分析表明,与SD转录组相比,ENR转录组中与锥体细胞类型相关的空间定位标记基因Nos1和Inhba比例升高,这与功能网络尺度上更高的LFP频率和振幅存在相关性(*** , ANOVA)。
通过皮尔逊相关系数 评估了交叉验证预测值与真实值之间的关系,该分析针对SRT-spot-n-Ephys电极进行(图6a和图S8,支持信息)。在对ENR环路上调表达的特异基因家族实施XGBoost算法后,我们观察到ENR数据集的预测精度显著提升。XGBoost分类器在ENR数据集上达到 的准确率,在SD数据集上为 (图6b)。这种转录组层面的单个基因或基因家族与网络层级功能之间的预测性相互作用,可能支持大脑功能是通过遵循基本组织原则的多尺度网络来协调的观点[20]。这些结果强调了神经活动、可塑性及特定基因家族内空间基因表达之间存在多尺度因果关联,这与先验经验调控网络层级功能的预测相符[17]。针对预测会影响特定n-Ephys活动特征的特殊基因,可通过定向操控来验证其功能作用[54]。这种方法能揭示塑造神经元响应及整体脑功能的关键基因与细胞通路,同时解析调控神经动力学、可塑性及疾病发病机制的分子机制。通过利用数据驱动学习,我们的模型该实现识别了转录组状态与功能网络动态之间的预测性关联,而无需强加严格的机械假设。虽然离子通道和突触调节剂等关键效应因子自然成为重要的预测指标,但模型能基于网络整体整合自主对转录组特征进行排序。展望未来,MEA-seqX为机制探索及基因-功能因果相互作用的实验验证提供了可扩展框架。我们的集成方法将大幅降低实验复杂性,从而增强结果可解释性,并为后续研究提供指导。
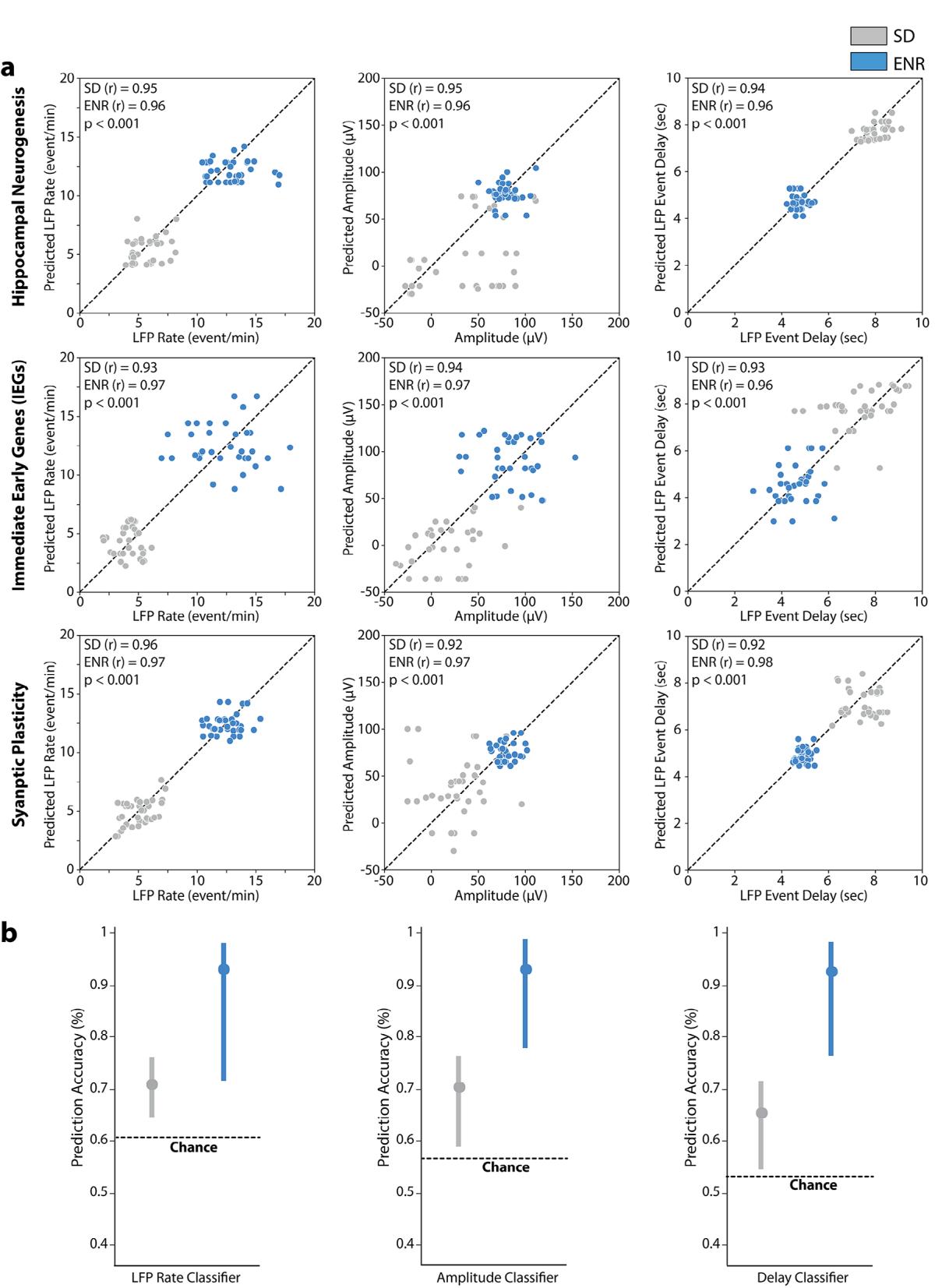
图6. 多尺度转录功能数据的机器学习预测。a) 应用XGBoost算法从SD和ENR数据中各指定基因家族的转录组数据预测网络电生理指标(LFP频率、振幅和LFP事件延迟)。通过皮尔逊相关系数®评估SD和ENR条件下从转录组数据预测n-电生理指标的效果。所有基因家族中预测的SD与ENR值之间存在显著差异 ,ANOVA)。b) 所有基因家族的最终数据输出迭代显示,XGBoost对ENR数据的预测精度高于SD数据,其性能通过平均准确度值比较得以体现。计算得出的均值及三个标准差被确定处于随机阈值范围内。
¶ 3. 结论与展望
本研究介绍了MEA-seqX平台,该平台具备前所未有的能力,可在完整脑组织中同时捕获并整合跨尺度的分子与功能信息。MEA-seqX通过融合多种技术优势并克服其各自局限,突破了Patch-seq、Electro-seq、CaRMA和空间转录组学等现有技术的边界。该平台利用CMOS-MEA技术的高密度特性,结合空间转录组学、光学成像与计算工具,为解决有限时空分辨率提供了独特方案,在空间语境中实现了卓越的时间分辨率。这一平台填补了关键认知空白,让我们得以理解支撑并解析生理性和经验依赖性可塑性范式中大规模神经交互完整性的分子基础设施。[20,55]通过整合性研究方法,我们识别了分子-功能特征间具有空间分辨率的因果调控规律,揭示了环境因素对协调性神经活动与基因表达的影响——这一关联虽被推测存在,但先前几乎无法验证。
该平台的图论分析揭示了一种具有密集连接枢纽复合体的小世界拓扑结构,表明存在分子功能专业化及跨尺度的全局通信能力提升[32,35,56]。此外,通过结合DPT与CAT分析,MEA-seqX在空间背景下沿发育轨迹追溯了细胞的伪时间排序及大规模神经活动进程,为海马回路中的协调时空动态提供了独特见解。MEA-seqX还展示了细胞类型识别的潜力,并凸显了海马回路中神经细胞类型的异质性及其网络范围内的频谱特征。该平台利用机器学习算法实现的预测能力,可基于空间基因表达精准预测全网络电生理活动特征,揭示了特定基因表达与神经活动之间的多尺度因果关系,从而为理解神经动力学提供新视角,这可能为机器学习与人工智能研究开辟新途径。将MEA-seqX与智能神经网络相结合,或能深化我们对复杂数据、决策过程及学习机制的理解[57,58]。
在高分辨率下整合分子与功能数据。虽然不用于直接监测患者,但MEA-seqX建立了患者特异性的分子和功能特征,为精准治疗策略提供依据。这种多尺度方法可提供患者特异性生物标志物的详细视图,有助于阿尔茨海默症、帕金森病和癫痫等复杂神经系统疾病的早期诊断及定制疗法的开发。MEA-seqX整合海量数据的能力使其成为精准医学的关键工具,能够识别不同组织类型的多尺度模式。通过结合机器学习和预测建模,该平台可预测疾病进展与治疗反应,实现动态的个性化治疗干预。这一能力与个性化医疗的最新进展相契合,强调整合多样化数据集以优化个体化患者护理[59]。 此外,MEA-seqX在药物研发领域潜力巨大,通过评估分子及全网络效应,为筛选和评估新药疗效提供综合系统。这种双重能力确保该平台可广泛应用于多种疾病及患者特定状况,推动个性化医疗解决方案的发展。尽管离体脑片制备会固有地破坏全脑长程连接,但MEA-seqX能有效捕捉局部和中尺度环路交互作用,实现高分辨率的功能-转录整合。海马脑片模型保留了关键突触通路和代偿性重组机制,便于研究网络动力学与适应性可塑性。尽管缺失远程投射,该平台仍能揭示局部环路在功能和转录层面的重组方式,为神经计算与疾病机制提供重要见解。本研究通过MEA-seqX探究长期环境富集引发的内源性全网络适应,但该平台保持严格时间数据对齐的能力,使其可轻松扩展至涉及急性扰动、药物干预或瞬时神经状态的研究领域。
展望未来,MEA-seqX技术存在多个提升潜力的发展机遇。技术与数据分析算法的持续进步有望进一步提高该平台的分辨率与预测精度。随着MEA-seqX与技术发展同步演进,其整合频域解析分析的能力可确保精准区分局部与全局功能关联。本研究通过将空间组织的局部场电位活动( )与特定海马亚区的转录组特征进行对齐,实现了介观尺度下功能数据与分子数据的整合。局部场电位作为可靠的功能读数载体,既能通过频域特异性洞察微环路活动,又保持着对大规模网络动态的检测灵敏度。这种宽频带检测既反映了群体层面的神经活动,又提供了足够的空间粒度以支撑稳健的多模态对应分析,且无需达到单细胞分辨率水平。未来研究可通过频带分离技术优化局部活动映射,更清晰地区分局部与全局功能关联,并随着排序算法的成熟及单细胞辨别精度与保真度的提升,引入尖峰解析分析方法。这些特性使MEA-seqX成为整合新兴技术的自适应平台采用高密度电极技术和单细胞分辨率转录组学。
整合蛋白质组学和表观基因组学数据等额外模态,可为神经信息处理提供更全面的研究视角[57]。MEA-seqX平台的适应性不仅限于神经组织,更可扩展至嗅球与心脏组织等电活性组织。这为研究不同背景下基因表达与电生理学的相互作用提供了可能。该技术在心脏组织中的应用有助于揭示心脏功能机制,推动心脏病学发展。
虽然目前的MEA-seqX实现利用了基于有源像素传感器技术构建的CMOS-MEA(4096电极)[14,17][60],但该平台的可扩展性超越了单一技术的局限。它能适配各种高密度技术,例如通过开关矩阵技术实现的(26400电极)[61]等系统。重要的是,MEA-seqX的应用范畴可突破离体研究的限制,延伸至更广阔的活体研究领域。通过与神经像素[62]、SiNAPS[63]等尖端活体探针或其他新兴模式的集成,该技术有望在活体生物中研究功能性神经动态与基因表达模式,从而架起实验室发现与真实生物情境之间的桥梁。
¶ 4. 实验部分
¶ 多尺度数据采集与分析工作流
为了全面概述多尺度数据采集与分析流程,图S1(支持信息)提供了详细的工作流程示意图。该工作流程从同一组织中的神经元电生理记录和空间转录组学技术数据采集开始,逐步展示了关键处理步骤。多尺度分析流程以阶梯式呈现,分别说明了nEphys数据和空间转录组数据的预处理、分析及特征提取过程。该工作流重点揭示了这些步骤如何最终实现分子与功能数据的整合,从而动态、多尺度地展现空间转录组与神经网络活动之间的因果关系。
¶ 动物与急性脑切片制备
所有实验均在12周龄C57BL/6J小鼠(查尔斯河实验室,德国)上进行,遵循适用的欧洲和国家法规(《动物保护法》),并获地方主管部门批准(萨克森州管理局;25-5131/476/14)。雌性C57BL/6J小鼠于五周龄时引入,随机分为两个实验组:标准饲养组(SD)和丰富环境饲养组(ENR),方法如前所述[17]。ENR组小鼠生活在特制笼具中,内含可重组玩具、迷宫式塑料管、隧道、栖居所及额外垫料。该ENR笼具环境被证实能通过增加刺激和差异化社交互动来促进经验依赖性可塑性的增强。小鼠在实验开始前于指定环境中适应六周,并持续饲养至实验当日。急性脑切片制备参照既往研究方案[14,17]。简言之,小鼠在断头前采用异氟烷麻醉,仔细取出脑组织后置于冰镇切片用蔗糖溶液中,随后进行切片处理。定制琼脂糖容器并固定于切割板,使用Leica VT1200S振动切片机(德国Leica Microsystems公司)制备背侧、水平及海马-皮层切片(厚度 )。切片在 条件下操作,溶液为含高浓度蔗糖的人工脑脊液(组分浓度单位 :250蔗糖、10葡萄糖、1.25磷酸二氢钠 、24碳酸氢钠 、2.5氯化钾、0.5抗坏血酸、4氯化镁 、1.2硫酸镁 、0.5氯化钙 ),持续通入 氧气 与 二氧化碳 (混合气,pH值维持在 。随后将海马-皮层切片在 环境下孵育45分钟,室温恢复至少1小时后,使用高密度神经芯片进行网络电生理记录。记录期间灌注液组分(mM):127氯化钠、2.5氯化钾、1.25磷酸二氢钠、24碳酸氢钠 、25葡萄糖、1.25硫酸镁,2.5 氯化,并通入钙 的氧气 和 二氧化碳
¶ 海马-皮质切片中细胞外n-Ephys记录
所有电生理记录均采用高密度CMOS生物传感微电极阵列芯片(CMOS-MEA)及定制化采集系统(3Brain AG,瑞士)完成。具体使用CorePlate 1 W 27/42 CMOS-MEA芯片,其配置4096个 微米2记录微电极,以 阵列排布,电极间距 微米,构成 平方毫米2有效传感区域。芯片内置放大电路支持 带通滤波,全局增益60 dB足以记录慢波与快振荡[14]。 进行细胞外记录时,使用定制铂丝压网将脑片转移并耦合至芯片表面。为减少实验变异并维持切片活性,采用热稳定灌流系统以4.5毫升/分钟−1流速持续输送氧合记录液,实验全程控温 。通过 微米4-氨基吡啶(4AP,德国Sigma-Aldrich)诱导药理激活响应,以 电极采样频率与 记录频率采集自发全网络活动信号,获取10分钟细胞外记录数据[17]。 所有溶液均现配现用,药理化合物溶解于记录液中使用。系统集成定制模块化体视显微镜(德国LeicaMicrosystems),可在进行高内涵纳米电生理记录时同步获取急性脑片光学成像。离线分析阶段,这些图像用于维持脑片组织空间结构与n-Ephys CMOS-MEA电极排布的对应关系。
¶ 海马-皮层切片中的空间分辨转录组学
采用Visium空间基因表达v1检测技术(10X Genomics, USA)对海马体进行空间分辨率测序和转录组分析。该类空间转录组学切片设有四个独立捕获区域,每个区域包含5000个直径 微米的空间条形码点,共同组成 毫米 毫米的捕获面积,足以容纳完整的小鼠海马体切片。选择n-Ephys与空间转录组学技术是基于其兼容的电极尺寸与空间点阵配置,可优化多模态整合的空间对应关系。在完成n-Ephys记录(10分钟)与光学成像(2分钟)后,立即将切片包埋于含OCT冷冻包埋剂的 毫米 毫米 Tissue-TEK冷冻模具中(1分钟),置于干冰速冻(1分钟),最后转移至WHEATON CryoELITE组织储存管进行低温保存。样本在 条件下保存以维持组织稳定性和活性,直至空间转录组实验日期。为优化每个位点的细胞数量并获得清晰的转录组图谱,使用赛默飞Cryostar NX70冷冻切片机将组织水平冷冻切片至 厚度。组织切片贴附于空间基因表达载玻片后,在 甲醇中固定30分钟。经苏木精-伊红染色后,通过明场成像获取形态学切片图像。成像完成后,切片在热循环仪中酶促透化22分钟,释放的mRNA与每个位点内数千个空间条形码标记的mRNA捕获寡核苷酸探针结合。 使用10X Genomics逆转录酶混合物在 热循环仪中孵育45分钟,将寡核苷酸结合的mRNA逆转录为cDNA。随后加入10X Genomics第二链合成混合物,于 热循环仪孵育15分钟完成cDNA第二链合成。为变性酶反应,添加pH 8.7的Qiagen EB缓冲液,最终样品按捕获区域分装至含Tris-HCl的对应试管中。通过PCR扩增制备具有空间条形码的全长cDNA文库。 取1μL各cDNA样本与KAPA SYBR FAST qPCR预混液加入洁净qPCR板,经孵育程序测定cDNA样本的Cq值为15.7,对应16个扩增循环。根据该Cq值向cDNA样本管添加10X Genomics扩增混合物完成qPCR扩增程序。测序前样本于 静置过夜。文库构建与测序在德累斯顿概念基因组中心完成,使用Illumina HiSeq 2000测序平台。测序数据通过10X Genomics Space Ranger流程处理,基于载玻片捕获区域边界的光学定位点将H&E染色明场图像与空间条形码基因表达数据对齐,执行组织定位、光学标记点识别及条形码/UMI计数。
¶ 数据分析
本研究采用的所有基础和高级算法均通过自行编写的Python脚本开发实现。为促进MEA-seqX平台的应用,完整脚本及示例数据集已发布于GitHub存储库(https://github.com/HayderAminLab/MEA-seqX)。所有使用的扩展包均已按规范引用。
¶ SRT质量控制与基因表达标准化
在数据分析之前,使用单细胞分析工具包Seurat[64]及附加工具STUtility(https://ludvigla.github.io/STUtility_web_site/)排除了技术批次效应和实验变异。这些工具包通过统计学方法量化了所有样本与条件下独特基因数量(nFeature RNA)和UMI数量(nCount RNA)。为深入解析并识别两种条件下共享的海马体结构,额外加载的Harmony包重新计算了UMAP嵌入与聚类分析,最终生成数据的集成低维表征[65]。经检测发现每个数据集均具有≈5500中位每个数据集中,每个位点的基因数量低于1000个独特基因的SRT位点被排除在分析之外。接着,为缩减分析所需的基因总数,线粒体和核糖体蛋白编码基因也被过滤掉。最后,为消除技术性批次变异并检测高变基因,每个SRT位点的总体基因表达通过各基因在所有SRT位点上的总计数进行标准化,使得标准化后每个位点具有相同的计数。这一步骤通过scanpy.pp.normalize_total Python包实现,代码可在GitHub(https://github.com/theislab/scanpy)获取。[66]
¶ 振荡模式检测与波形分类
在数据分析之前,我们采用硬阈值算法对每次记录中的局部场电位振荡模式进行检测[17]。此外,检测到的事件会通过低通四阶巴特沃斯滤波器( )进行进一步处理和滤波。最后,通过自定义编写的Python脚本运用分位数阈值法,剔除异常放电电极或非生理性检测事件[17]。为表征记录的LFP振荡波形特征并将其归因于海马互连层次结构,我们按照既往方法采用主成分分析和K均值聚类算法对波形特征进行系统性归类[14]。
¶ 结构簇
为了表征局部和全局海马子网络行为,研究人员通过将光学显微镜海马图像叠加在CMOS微电极阵列布局上,使功能性放电神经电生理电极与特定海马区域建立结构关联。随后根据海马切片上的结构标记——齿状回、门区、CA1区、CA3区、内嗅皮层和梨状皮层,将电极分组形成集群[17]。为解析这六个主要区域的转录组特征,通过使用Loupe Browser(10X Genomics, 美国)将H&E染色明场显微镜图像叠加在空间转录组点位布局上,使SRT点位与特定海马区域形成结构对应。
¶ 多尺度空间对齐
为了推断n-Ephys电极-SRT位点界面与其各自网络范围内的功能性电活动及具有空间定位的转录组特征读数之间的对应关系,MEA-seqX实施了多尺度空间配准程序。为在空间背景下提供同一网络的转录组和电生理图谱,我们通过图像尺寸调整和旋转实现了自动切片配准。该配准基于光学成像、n-Ephys电极-SRT位点界面的物理尺寸及相关海马-皮层结构输入,将多尺度数据置于同一维度中。首先,MEA-seqX基于n-Ephys电极-SRT位点尺寸实现自动缩放算法,将SRT的亮视野显微镜切片H&E染色图像尺寸调整至n-Ephys的相应海马区光学显微镜图像。值得注意的是,由于技术分辨率差异,n-Ephys电极与SRT位点的匹配并非一一对应,而是基于相关海马-皮层结构输入的分数匹配。因此,每个SRT位点会关联若干n-Ephys电极,并获取来自相关电极的平均电生理特征。接着,通过以下步骤计算两个切片图像间的空间配准与旋转:i)分别为SRT和n-Ephys两个尺度分配海马-皮层结构参考点{i, j}与{k, l},其中{i, k}为齿状回脊的中点,{j, l}位于齿状回上叶片边缘;ii)将SRT参考点i与n-Ephys参考点 i) 使用参考点k将两个标度置于同一维度。ii) 在参考点 完成对齐后,基于 之间的差异对参考点 进行最终对齐。iv) 在已知两个阵列坐标的前提下,通过 、 、y和y′的距离计算 的夹角。v) 为确定 ,采用对齐参考点 形成的水平交线定义 ,同时通过参考点j与该水平交线的垂直交线定义 。vi) 为确定 ,采用对齐参考点 }形成的水平交线定义 ,同时通过参考点l与该水平交线的垂直交线定义 。vii) 最终旋转角度定义为 ,该角度应用后可使参考点 对齐为最终多标度参考点 ,并使参考点 对齐为最终多标度参考点 。
¶ 功能性网络平均活动特征
为了确定空间基因表达模式如何与功能性神经电生理特征相关联,我们使用斯皮尔曼相关系数[25]将筛选出的基因与某一网络特征进行关联分析,并依据本杰米尼-霍克伯格错误发现率校正后的p值[67]按显著性进行排序。大规模时空局部场电位振荡的功能性网络活动特征包括:LFP发放率、振幅、能量、LFP事件延迟以及正负峰值计数[14,17]。
¶ 靶向基因列表
基于功能基因本体论,制定了特定的基因列表。与即刻早期基因家族、信号通路、海马功能和神经发生相关的基因被汇编成六个列表:即刻早期基因、海马神经发生、海马信号通路、受体和通道、突触可塑性、突触小泡和粘附。[26,27]
¶ 非负矩阵分解
采用稀疏约束非负矩阵分解的无监督机器学习算法,用于识别从SRT和n-Ephys网络中出现的个体时空模式。[28] NMF分解法的描述基于Scikit-learn 1.2.2 Python工具包(sklearn.decomposition.NMF)并进行了适应性调整。[68] 首先,输入V矩阵包含SRT基因表达值(即IEG家族)与n-Ephys活动特征(即LFP速率)或拓扑指标(即度)相关联的集体信息。每个数据条目包含与网络活动特征或拓扑指标值相关的各基因表达值 及其空间定位信息 ( m ) _
其中 是空间局部化斑点, 是基因表达与网络特征或指标的相关性, 是因素数量。
结果分解基础W矩阵包含因子权重贡献的时空分布(n-电生理特征或度量)。系数H矩阵表示基因表达模式对每个空间位置和n-电生理特征或度量剖面的推断贡献。为了优化V与乘积矩阵H和W之间的距离,广泛使用的距离优化函数平方Frobenius实现了范数(F),这为因子添加了稀疏性约束。[68]
其中 和 是非负值( )。 和 分别是 和 W 对应的正则化参数。
¶ 互信息
为了呈现基于基因表达的多层网络中点的集体性,我们计算了每个目标基因列表的基因表达分布。接着,计算了每个点中各目标基因列表的基因信息的互信息距离分数,在点之间进行比较,并按聚类排序。[30] 互信息的计算使用了Scikit-learn 1.2.2 Python包中的适配函数(sklearn.metrics.normalized_mutual_info_score)。[68]
¶ 功能性连接性
为了推断多层海马网络中功能放电活动的大规模统计依赖连接性,我们采用先前描述的方法,计算了成对放电神经电生理电极间的互协方差:先使用皮尔逊相关系数,再结合定向传递函数和多元格兰杰因果分析。[14,17]
¶ 图谱可视化
为了可视化SRT和n-Ephys数据集的大规模网络连接性,我们分别基于互信息距离分数和功能连接数据结构构建了包含节点与边的网络模型,方法如前所述[17]。数据被转换为(.gexf)文件格式,并直接通过Gephi 9.2版本软件(https://gephi.org)进行读取和可视化。为研究选定基因在空间网络阵列中的功能交互作用,我们筛选了所有配对点之间的互信息分数,仅保留超过均值两个标准差以上的数据。因此设定阈值,使每个目标基因列表中的基因包含 的关联强度。针对n-Ephys电极的功能连接分析,我们纳入了总功能连接中排名前 的链接。SRT与n-Ephys连接图谱均采用相似的边权重和度范围查询进行绘制。
¶ 网络拓扑度量
采用图论来基于n-Ephys检测到的LFP事件的功能连接性或SRT基因表达中的互信息分数,表征整体网络拓扑和互联性。拓扑指标通过自定义Python代码进行筛选,如先前报道[14,17]。简言之,网络连接拓扑指标通过将节点 视为图中可能相互连接或不相连的核心组件来描述。在这种情况下,节点n对应于传感阵列中的特定n-Ephys电极或SRT捕获点,而边e是每个节点n之间的功能链接或连接。为呈现整体网络拓扑和特征,选取了以下图论拓扑参数:
Degree为了表征网络连通性的不同表现形式,节点的度k被用来描述节点n的特性。
描述连接到一个节点的边的数量如前所述。[17]
其中 表示节点i的度。 表示节点i和j之间的连接关系。N是网络中所有计算节点的集合。
¶ 枢纽节点与核心枢纽群
为确定网络中的中心化重要节点并揭示网络拓扑结构,本研究对枢纽节点和富人俱乐部节点进行了分析。枢纽节点的识别基于三项节点指标——节点强度、聚类系数和网络效率。通过计算各节点的指标值并比对,判定节点是否处于所有节点前 的数值区间[17]。为严格界定枢纽节点,采用枢纽评分设定限制条件:该评分取值0-3分,对应节点在三项指标中进入前 的数量(0项、1项、2项或全部3项),最终将满足至少两项标准的节点定义为枢纽节点。在枢纽节点群中存在一个连接密集的子群,即富人俱乐部节点,这类节点被描述为具有高于平均程度的枢纽节点,其特性通过富人俱乐部系数 予以量化。
其中k表示度, 代表度数大于给定值k的节点数量,而 表示由 组成的子网络中的连接数。
¶ 网络拓扑特征分析
为了确定枢纽节点对网络功能以及塑造网络拓扑的组织过程的潜在影响,在n-Ephys和SRT数据集中对检测到的节点的度分布 进行了表征,这导致了具有幂律尾的衰减分布[34]。
为了估算描述具有小世界属性的无标度拓扑的幂律度分布 ,研究采用了对数正态模型拟合。
其中 和 分别表示该分布的均值和标准差。为可视化最优拟合的网络特征,我们采用互补累积分布函数而非节点度的概率密度,并采用对数坐标轴绘制,以便更稳健地呈现高k值区间的特征。通过实际数据与拟合模型之间的拟合优度检验,并以决定系数 进行评估。最后应用帕累托线性分箱法(scipy.stats.pareto)[69]对幂律分布进行离散化处理。
¶ 扩散伪时间
为从静态、空间分辨的测序数据中精准定位动态转录变化,并确定内在与外在因素对特定动态过程的影响,本研究采用了扩散伪时间(DPT)方法。[38] DPT技术能揭示生物过程的内在动态机制,在本案例中,它通过空间分辨的海马转录组数据,解析了特定基因表达的时间轨迹。简言之,DPT通过在降维扩散图空间中将转录状态转换建模为扩散过程,重建伪时间轨迹,并根据各空间转录组点向特定分子状态演变的概率顺序进行排序。这种伪时间推断基于基因表达状态在空间中的相似性重建潜在转录进程,而非实时观测。传统扩散图虽能有效降噪并保持局部与全局结构,但生成的图谱通常以高维形式编码信息,限制了可视化效果。为在DPT分析前克服此局限,本文实现了基于热扩散的亲和力转换嵌入(PHATE)技术在空间转录组数据中的应用,该方法可在低维度呈现信息(https://github.com/KrishnaswamyLab/PHATE)。[70] 该技术通过势距离编码全局数据关联性——将局部相似性转化为转移概率。这些扩散概率通过将局部信息转化为随机游走单步转移概率来确定,可进行t步幂运算得到局部与全局距离的t步游走概率。在本数据集中,每个空间位点通过加权图与最近邻或远端位点建立确定的关联关系[70],DPT分析则根据位点向不同分化方向演变的概率对转录组位点进行排序[38]。
¶ 细胞类型解卷积
为了确定细胞类型共定位并检验两个海马转录组之间的差异,我们采用单细胞测序参考数据进行了CARD分析。[45,46]基于CARD的分析方法已发布在GitHub平台(https://github.com/YingMa0107/CARD),并针对Python环境的空间转录组数据进行了适配。该参考数据包含对海马细胞类型的广泛类别划分,涵盖星形胶质细胞、内皮细胞、室管膜细胞、巨噬细胞、小胶质细胞、神经源性细胞、神经元、少突胶质细胞和多突胶质细胞及其对应亚群。通过低计数细胞类型筛选,海马细胞类型从85种缩减至76种。
¶ 基于XGBoost算法的预测
为了确定特定基因表达值是否能够预测每个SRT位点的相关HC网络特征参数(如局部场电位频率、振幅和LFP事件延迟),我们采用了梯度提升(XGBoost)算法。该算法整合了多个树模型并具有强可解释性[53]。实现过程使用Scikit-learn 1.2.2 Python软件包函数(sklearn.ensemble.GradientBoostingClassifier与sklearn.model_selection.train_test_split)[68]。输入数据集包含预定义基因列表的空间解析基因表达值,以及功能性n-Ephys数据中的相关网络特征。为使SD与ENR数据集在输入表征上具有可比性,鉴于ENR数据集网络特征参数存在固有双倍差异(如先前所述),我们对其进行了随机二次采样处理,保留半数数据[17]。
数据集被划分为训练集和测试集( 训练, 测试;采用sklearn.model_selection.train_test_split方法),模型经过100次迭代训练。随后将预测输出值与真实数据点进行比较,以评估从转录组数据预测网络特征的可预测性、准确性和显著性[71]。用于统计分析的软件包包括:Scikit-learn 1.2.2 Python包(用于计算预测准确度sklearn.metrics.explained_variance_score)[68],以及Scipy 1.10.1(用于计算皮尔逊相关系数scipy.stats.pearsonr和scipy.stats.ttest_ind)[69]。预测准确度通过多个最终数据输出进行验证,将超过平均值三个标准差的值定义为处于随机概率阈值范围内。训练完成后,只要保持相同的基因列表(或列表中的单个基因)和电生理特征,XGBoost模型即可自主根据空间转录组数据预测电生理特征,无需额外人工输入。然而当引入新基因列表、不同电生理指标或改变实验范式(如不同脑区、条件或疾病状态)时,模型将通过内置划分方法( 训练, 测试)自动重新训练。这种自适应机制确保了处理新增或修改输入数据时能获得最优且准确的预测结果。
¶ 统计分析
所有统计分析均使用Originlab 2020或按插件附加说明进行操作。除非特别标注为标准差,本研究中数据均以均值 平均标准误(SEM)表示。箱形图由第25至第75百分位数确定,须线表示第5至第95百分位数(长度限于四分位距的1.5倍范围内)。图中线条代表中位数,方块标记代表均值。组间差异的统计学显著性检验(如适用)采用Kolmogorov-Smirnov检验、单因素方差分析(ANOVA)或双因素方差分析结合Tukey事后检验。 <小于0.05视为具有统计学意义。
¶ 数据可用性声明
支持本研究结果的所有数据均在正文及图表中提供。此外,数据样本及方法相关的Python脚本可在我们实验室的GitHub存储库(https://github.com/HayderAminLab/MEA-seqX)获取。其中包括存储于Zenodo的测序数据(访问地址:https://doi.org/10.5281/zenodo.10626259),以及网络电生理LFP(n-Ephys)数据(访问地址:https://doi.org/10.5281/zenodo.10620559)。
¶ 关键词
人工智能机器学习、连接组、经验依赖性可塑性、大规模神经记录、预测建模、空间转录组学、时空动态
收到日期:2024年10月4日修订日期:2025年4月4日在线发布日期:2025年4月30日
[1] R·弗拉科维亚克、H·马克拉姆,Philos. Trans. R. Soc., B 2015年,370,20140171。
[2] I·帕伦蒂、L·G·拉巴内达、H·舍恩、G·诺瓦里诺,Trends Neurosci. 2020年,43, 。
[3] J·J·帕洛普、L·穆克,Nat. Rev. Neurosci. 2016年,17,777。
[4] C·R·卡德威尔、A·帕拉桑察、X·姜、P·贝伦斯、Q·邓、M·耶尔马兹、J·赖默、S·沈、M·贝特格、K·F·托利亚斯、R·桑德伯格、A·S·托利亚斯,Nat. Biotechnol. 2016年,34, 。
[5] J·富齐克、A·蔡塞尔、Z·马特、D·卡尔维吉奥尼、Y·柳川、G·萨博、S·林纳尔松、T·哈卡尼,Nat. Biotechnol. 2016年,34,175。
[6] 李琪、林钊、刘睿、唐旭、黄健、何毅、隋昕、田雯、沈晖、周航、盛华、史慧、肖磊、王旭、刘健,Cell 2023年,186,2002。
[7] 徐帅、杨皓、V·梅农、A·L·勒米尔、王玲、F·E·亨利、S·C·图拉加、S·M·斯特恩松,Science 2020年,370,605。
[8] P·L·斯塔尔、F·萨尔门、S·维科维奇、A·伦德马克、J·F·纳瓦罗、J·马格努松、S·贾科梅洛、M·阿斯普、J·O·韦斯特霍尔姆、M·胡斯、A·莫尔布林克、S·林纳尔松、S·科德卢皮、Å·博里、F·庞滕、P·I·科斯泰亚、P·萨伦、J·穆尔德、O·伯格曼、J·伦德伯格、J·弗里森,Science 2016年,353, 。
[9] L·拉尔松、J·弗里森、J·伦德伯格,Nat. Methods 2021年,18,15。
[10] S·马尼亚蒂斯、T·艾约、S·维科维奇、C·布雷恩、K·康、A·莫尔布林克、D·法格加尔捷、 ·安德里西科娃、S·萨伦佩、G·赛斯-卡斯特罗、M·奎瓦斯、A·沃特斯、J·伦德伯格、R·博诺、H·法特纳尼,Science 2019年,364,89。
[11] M·阿斯普、S·贾科梅洛、L·拉尔松、C·吴、D·富尔特、X·钱、E·沃德尔、J·库斯托迪奥、J·赖梅高、F·萨尔门、C·奥斯特霍尔姆、P·L·斯塔尔、E·桑德斯特伦、E·奥克松、O·伯格曼、M·比恩科、A·曼森-布罗伯格、M·尼尔松、C·西尔文、J·伦德伯格,Cell 2019年,179,1647。
[12] A. E. Urai、B. Doiron、A. M. Leifer、A. K. Churchland,Nat. Neurosci. 2022,25,11.
[13] G. Buzsáki,Nat. Neurosci. 2004,7,446.
[14] X. Hu、S. Khanzada、D. Klütsch、F. Calegari、H. Amin,Biosens. Bioelec- tron. 2022,198,113834.
[15] H. Amin、T. Nieus、D. Lonardoni、A. Maccione、L. Berdondini,2017,7,13.
[16] K. Imfeld、S. Neukom、A. Maccione、Y. Bornat、S. Martinoia、P.-A. Farine、M.Koudelka-Hep、L. Berdondini,IEEE Trans. Biomed. Eng. 2008,55,2064.
[17] B. A. Emery、X. Hu、S. Khanzada、G. Kempermann、H. Amin,Biosens. Bioelectron. 2023,237,115471.
[18] B. A. Emery、X. Hu、L.Maugeri、S. Khanzada、D. Klütsch、H. Amin,IEEE EMBS 2022,42,4.
[19] B. A. Emery、S. Khanzada、X. Hu、D. Klütsch、H. Amin,J. Visualized Exp. 2024,205,66473.
[20] L. Luo、E. M. Callaway、K. Svoboda,Neuron 2018,98,256.
[21] S. Panzeri、J. H. Macke、J. Gross、C. Kayser,Trends Cognit. Sci. 2015,19,162.
[22] G. Kempermann,Nat. Rev. Neurosci. 2019,20,235.
[23] X. Hu、B. A. Emery、S. Khanzada、H. Amin,Front. Bioeng. Biotechnol. 2024,12,1.
[24] L. McInnes、J. Healy、J. Melville,arXiv.stat.ML 2018,https://doi.org/10.48550/arXiv.1802.03426
[25] K. M. Anderson、F. M. Krienen、E. Y. Choi、J. M. Reinen、B. T. T. Yeo、A. J. Holmes,Nat. Commun. 2018,9,14.
[26] J.Zhang、J. Jiao,Biomed Res. Int. 2015,2015,727542.
[27] L. M. Valor、P. Charlesworth、L. Humphreys、C. N. G. Anderson、S. G. N. Grant,Proc. Natl. Acad. Sci. USA 2007,104,4658.
[28] J. P. Brunet、P. Tamayo、T. R. Golub、J. P. Mesirov,Proc. Natl. Acad. Sci. USA 2004,101,4164.
[29] G. W. Milligan、M. C. Cooper,Psychometrika 1985,50,159.
[30] I. Priness、O. Maimon、I. Ben-Gal,BMC Bioinf. 2007,8,111.
[31] M. P. van den Heuvel、O. Sporns,J. Neurosci. 2011,31,15775.
[32] J. J. McAuley、L. Da Fontoura Costa、T. S. Caetano,Appl. Phys. Lett.2007,91,2.
[33] D. J. Watts、S. H. Strogatz,Nature 1998,393,440.
[34] A.-L. Barabási、R. Albert,Science 1999,286,509.
[35] R. W. Overall、G. Kempermann,Front. Neurosci. 2018,12,641
[36] A. Arnatkeviciute、B. D. Fulcher、S. Oldham、J. Tiego、C. Paquola、Z. Gerring、K. Aquino、Z. Hawi、B. Johnson、G. Ball、M. Klein、G. Deco、B. Franke、M. A. Bellgrove、A. Fornito,Nat. Commun. 2021,12,4237.
[37]A. M. Zador、J. Dubnau、H. K. Oyibo、H. Zhan、G. Cao、I. D. Peikon,PLoS Biol. 2012,10,1001411.
[38] L. Haghverdi、M. Büttner、F. A.Wolf、F. Buettner、F. J. Theis,Nat. Meth- ods 2016,13,845.
[39] Z.C. Chao、D. J. Bakkum、S. M. Potter,J. Neural Eng. 2007,100,294.
[40] M. Gandolfo、A. Maccione、M. Tedesco、S. Martinoia、L. Berdondini,J. Neural Eng. 2010,7,056001.
[41] W. Zhang、D. J. Linden,Nat. Rev. Neurosci. 2003,4,885.
[42] J. Richiardi、A. Altmann、A. C.Milazzo、C. Chang、M. M. Chakravarty、T. Banaschewski、G. J. Barker、A. L. W. Bokde、U. Bromberg、C. Büchel、P. Conrod、M. Fauth-Bühler、H. Flor、V. Frouin、J. Gallinat、H. Garavan、P. Gowland、A. Heinz、H. Lemaître、K. F. Mann、J. L. Martinot、F. Nees、T. Paus、Z. Pausova、M. Rietschel、T. W. Robbins、M. N. Smolka、R. Spanagel、A.Ströhle、G. Schumann等,Science 2015,348,1241.
[43] M. Hübener、T. Bonhoeffer,Neuron 2010,67,363.
[44] N. W. Gouwens、S. A. Sorensen、J. Berg、C. Lee、T. Jarsky、J. Ting、S. M. Sunkin、D. Feng、C.A. Anastassiou、E. Barkan、K. Bickley、N. Blesie、T. Braun、K. Brouner、A. Budzillo、S. Caldejon、T. Casper、D. Castelli、P. Chong、K.Crichton、C. Cuhaciyan、T. L. Daigle、R. Dalley、N. Dee、T. Desta、S. L. Ding、S. Dingman、A. Doperalski、N. Dotson、T. Egdorf等,Nat. Neurosci. 2019,22,1182.
[45] 马 Y.,周 X.,Nat. Biotechnol. 2022年,40, 。
[46] 凯布尔 D.M.,默里 E.,邹 L. S.,戈埃瓦 A.,马科斯科 E. Z.,陈 F.,伊里扎里R. A.,Nat. Biotechnol. 2022年,40,517。
[47] 欧文 S. R.,孙 W.,科普兰 M.,林多 S.,斯普拉斯顿 N.,塞姆布罗夫斯基 M. S.,Cell Rep. 2020年,31,107551。
[48] 克劳斯伯格 T.,索莫吉 P.,Science 2008年,321,53。
[49] 皮尼亚泰利 M.,瑞安 T. J.,罗伊 D. S.,洛维特 C.,史密斯 L. M.,穆拉利达尔 S.,利根川进,Neuron 2019年,101,274。
[50] 斯坦纳特 J. R.,切尔诺娃 T.,福赛思 I. D.,Neuroscientist 2010年,16,435。
[51] 邦 C. L. M.,加思韦特 J.,J. Neurosci. 2003年,23,1941。
[52] 奥伯伦德 K.,维特 V.,马利恩 A. S.,加斯 P.,本特森C. P.,巴丁 H.,Learn. Mem. 2022年,29,55。
[53] 李 W.,尹 Y.,权X.,张 H.,Front. Genet. 2019年,10,1077。
[54] 阿斯普 M.,贝里恩斯特罗勒 J.,伦德伯格 J.,BioEssays 2020年,42,1900221。
[55] 摩尔H.,莱加 B. C.,康普卡 G.,Curr. Opin. Cell Biol. 2022年,78,102118。
[56] 何 B. J.,Trends Cognit. Sci. 2014年,18,480。
[57] 塞诺夫斯基T. J.,丘奇兰 P. S.,莫夫松 J. A.,Nat. Neurosci. 2014年,17, 。
[58] 西格尔 M.,唐纳 T. H.,恩格尔 A. K.,Nat. Rev. Neurosci. 2012年,13,121。
[59] 何 A. Z. D.,夸克 S. R.,麦凯布 E. R. B.,庄 W. J.,周 E. K.,丁 X.,盖尔布 B. D.,金斯伯格 G. S.,哈森斯塔布 J.,何 C.-M.,莫布里 W. C.,诺兰 G. P.,罗森 S. T.,Trends Biotechnol. 2019年,38,497。
[60] 贝东迪尼 L.,伊姆费尔德 K.,马乔内 A.,泰德斯coM.,诺伊科姆 S.,库德尔卡-赫普 M.,马蒂诺亚 S.,Lab Chip 2009年,9,2644。
[61] 赫尔 F.,哈菲佐维奇 S.,乌格尼文科 T.,弗雷 U.,弗兰克斯 W.,佩里亚尔 E.,佩里亚尔 J.-C.,布劳 A.,齐格勒 C.,希尔勒曼 A.,Biosens. Bioelectron. 2007年,22, 。
[62] 君 J. J.,斯坦梅茨 N. A.,西格尔 J. H.,登曼 D. J.,鲍扎 M.,巴巴里茨 B.,李 A.K.,阿纳斯塔西乌 C. A.,安德烈 A.,阿伊丁 Ç.,巴比奇 M.,布兰奇T. J.,博宁 V.,科托 J.,杜塔 B.,格拉蒂 S. L.,古特尼斯基 D. A.,霍伊泽 M.,卡什 B.,莱多霍维奇 P.,洛佩兹 C. M.,米特鲁特 C.,穆萨 S.,奥昆 M.,帕奇塔留 M.,普策斯 J.,里奇 P. D.,罗桑特 C.,孙 W. L.,斯沃博达 K. 等,Nature 2017年,551, 。
[63] 安戈齐 G.N.,博伊 F.,勒孔特 A.,米勒 E.,马勒巴 M.,祖卡 S.,卡西莱 A.,贝东迪尼 L.,Biosens. Bioelectron. 2019年,126, 。
[64] 郝 Y.,郝S.,安德森-尼森 E.,莫克 W. M.,郑 S.,巴特勒 A.,李 M. J.,威尔克 A. J.,达比 C.,扎格 M.,霍夫曼 P.,斯托克修斯 M.,帕帕莱克西E.,米米图 E. P.,贾恩 J.,斯里瓦斯塔瓦 A.,斯图尔特 T.,弗莱明 L. M.,杨 B.,罗杰斯 A. J.,麦克尔拉思 J. M.,布利什 C. A.,戈塔尔多 R.,斯迈伯特 P.,萨蒂贾 R.,Cell 2021年,184,3573。
[65] 科尔松斯基 I.,米勒德 N.,范 J.,斯洛维科夫斯基 K.,张 F.,魏 K.,巴格拉延科 Y.,布伦纳 M.,罗 P.-R.,雷乔杜里 S.,Nat. Methods 2019年,16,1289。
[66] 沃尔夫 F. A.,安格勒 P.,泰斯 F. J.,Genome Biol. 2018年,19,15。
[67] 本杰明尼 Y.,霍赫贝格 Y.,J. R. Stat. 1995年,57, 。
[68] 佩德雷戈萨 F.,瓦罗坎 A.,格兰福特 A.,米歇尔 V.,蒂里翁 B.,格里塞尔 O.,布隆代尔 M.,普雷滕霍费尔 P.,韦斯 R.,杜堡 V.,范德普拉斯 J.,帕索斯 A.,库尔纳普 D.,布鲁彻 M.,佩罗 M.,杜谢奈 E.,JMLR 2011年,12,2825。
[69] 维塔宁 P.,戈默斯 R.,奥利芬特 T. E.,哈伯兰 M.,雷迪 T.,库尔纳普D.,布罗夫斯基 E.,彼得森 P.,韦克瑟 W.,布莱特 J.,范德瓦尔特 S. J.,布雷特 M.,威尔逊 J.,米尔曼 K. J.,马约罗夫 N.,纳尔逊 A. R.J.,琼斯 E.,克恩 R.,拉尔森 E.,凯里 C. J.,波拉特 I.,冯 Y.,摩尔E. W.,范德普拉斯 J.,拉克萨尔德 D.,佩尔克托尔德 J.,西姆尔曼 I.,亨里克森 I.,金特罗 E. A.,哈里斯 C. R. 等,˙ 2020年,Nat. Methods,261。
[70] 穆恩 K. R.,范迪杰克 D.,王 Z.,吉甘特 S.,伯克哈特 D. B.,陈 W. S.,伊姆 K.,范登埃尔曾 A.,希恩 M. J.,科伊夫曼 R. R.,伊万诺娃 N. B.,沃尔夫 G.,克里希纳斯瓦米 S.,172019年,Nat. Biotechnol.,1482。
[71] 基科 D.,沃伦斯 M. J.,尤尔曼G.,37 2021年,PeerJ Comput. Sci.,623。