¶ Immunity
¶ 小胶质细胞中的Trem2表达需要在发育过程中维持正常的神经元生物能量学
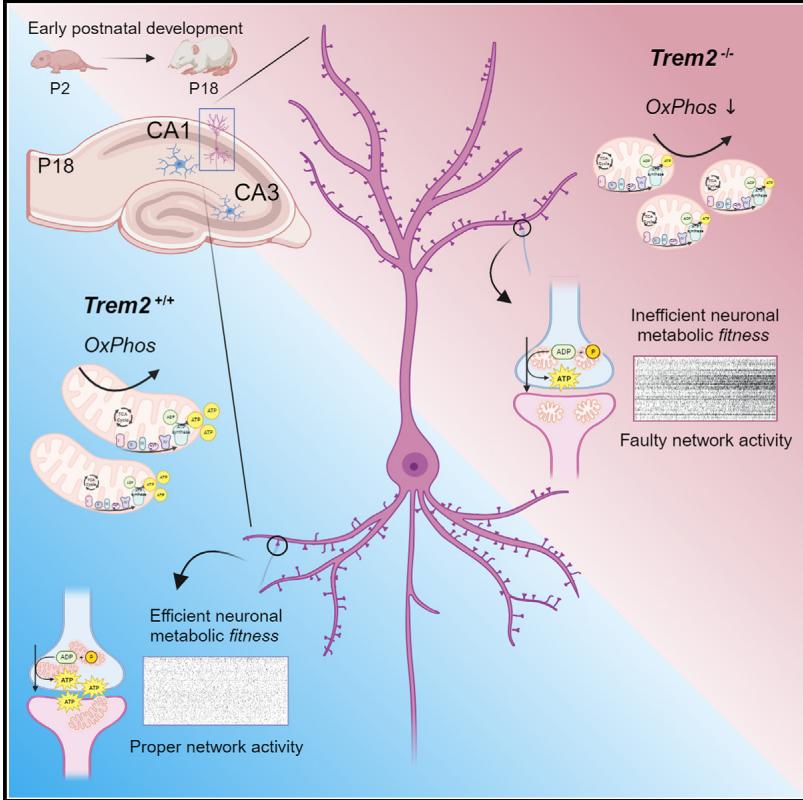
图形摘要
¶ 简而言之
Trem2对小胶质细胞介导的突触重塑至关重要,但Trem2是否参与调控神经元发育仍不明确。Tagliatti与Desiato团队研究表明,小胶质细胞中的Trem2以区域特异性方式调控海马神经元的代谢适应性。早期发育阶段小胶质细胞与神经元之间Trem2介导的通讯缺失,会将代谢紊乱与突触异常及神经回路成熟障碍联系起来。
¶ 亮点
小鼠中Trem2缺乏在发育期间损害海马神经元生物能量学
CA1区神经元而非CA3区神经元表现出线粒体质量和新陈代谢的降低
CA1区代谢功能障碍随后伴随出现突触和网络改变
Trem2 的部分减少足以改变神经元代谢适应性
¶ 小胶质细胞中的Trem2表达需要在发育过程中维持正常的神经元生物能量学
埃里卡·塔利亚蒂,1,2,8 詹尼·德夏托,1,8 萨拉·曼奇内利,3 马泰奥·比佐托,1,3 玛丽亚·C·加利亚尼,4 埃莉萨·法吉亚尼,1 丽贝卡·埃尔纳´德斯-索托,1 安德烈亚·库古拉,1 保拉·波利塞诺,1 马泰奥·米奥托,1 拉斐尔·J·阿圭略,5 法比亚·菲利佩洛,1,6 卡蒂亚·科尔特塞,4 拉法埃拉·莫里尼,1 西蒙娜·洛达托,1,3 与米凯拉·马泰奥利1,7,9,*
1 IRCCS Humanitas 研究医院,意大利米兰罗扎诺曼佐尼街56号,邮编20089
2 临床与实验癫痫科,UCL 女王广场神经学研究所,英国伦敦大学学院,英国伦敦
3 Humanitas 大学,生物医学科学系,意大利米兰皮耶韦埃马努埃莱莱维蒙塔尔奇尼街4号,邮编20072
4 细胞电子显微镜实验室,实验医学系(DIMES),人体解剖学,热那亚大学`,意大利热那亚安东尼奥·德托尼街14号,邮编16132
5 艾克斯-马赛大学,法国国家科学研究中心,法国国家健康与医学研究院,CIML,马赛-吕米尼免疫学中心,法国马赛
6 精神病学系,华盛顿大学医学院,美国密苏里州圣路易斯市
7 神经科学研究所 - 国家研究委员会,意大利米兰20139
8 这些作者贡献相同
9 主要联系人 *通讯作者:michela.matteoli@hunimed.eu https://doi.org/10.1016/j.immuni.2023.12.002
¶ 摘要
髓样细胞表达的触发受体2(Trem2)是一种在脑小胶质细胞中表达的髓系细胞特异性基因,其变异与神经退行性疾病(包括阿尔茨海默病)相关。Trem2对小胶质细胞介导的突触重塑至关重要,但Trem2是否参与调控神经元发育仍不明确。本研究揭示Trem2在发育过程中对锥体神经元生物能量谱调控的关键作用。缺乏Trem2时,海马角(CA)1区(而非CA3区)发育中的神经元表现出能量代谢受损,伴随线粒体质量减少和细胞器超微结构异常。与之相对应的是,出生时海马锥体神经元发生转录重编程,表现为代谢、氧化磷酸化和线粒体基因特征的广泛改变,同时CA1神经元成熟延迟。我们的结果揭示了Trem2通过以区域特异性方式调控神经元代谢适应性,在控制神经元发育中发挥重要作用。
¶ 引言
过去几年间,关于大脑是免疫特权器官的陈旧观念已被新发现取代——研究表明神经系统与免疫系统持续进行着相互作用,尤其在发育和衰老阶段更为显著。小胶质细胞作为大脑常驻的主要免疫细胞,在这些过程中扮演着关键角色。除了构成抵御病原体侵袭的防线外,小胶质细胞还被发现深度参与调节中枢神经系统正常发育与可塑性的生理功能,包括调控神经元凋亡、神经发生、髓鞘形成,以及在发育过程中清除多余突触等重要进程。1–9
髓系细胞表达的触发受体2(Trem2)是一种免疫球蛋白超家族跨膜受体,在大脑中仅由小胶质细胞表达10–15,它通过调控小胶质细胞的能量代谢来控制其功能性表现16–20。当与膜结合或可溶性配体(包括脂质、脂蛋白、DNA和细菌产物)通过衔接蛋白DAP10和DAP12激活Trem2的细胞内信号转导,增强小胶质细胞对凋亡神经元、细胞碎片、细菌产物及蛋白聚集体(包括神经毒性β-淀粉样肽)的吞噬作用。此外,Trem2能促进髓系细胞存活与增殖21–25,并调节炎症信号传导,控制小胶质细胞从稳态向疾病相关状态(DAM)转变26。膜结合型Trem2经α-分泌酶ADAM10和ADAM17切割27,28,或通过可变剪接的Trem2转录本翻译29,可释放具有生物活性的可溶性Trem2(sTrem2)至细胞外环境30–32。全基因组关联研究证实,Trem2错义纯合与杂合变异与神经退行性疾病(特别是阿尔茨海默病)相关33–37。具体而言,Trem2变体剥夺了小胶质细胞所需的专门感觉机制,使其无法检测中枢神经系统内的损伤,同时阻止它们获得对抗病理状态所需的转录和功能特征。38,39 因此,在AD模型或脱髓鞘范式中,Trem2的缺乏导致淀粉样斑块周围的神经突变性和轴突损伤增加。18,40–42
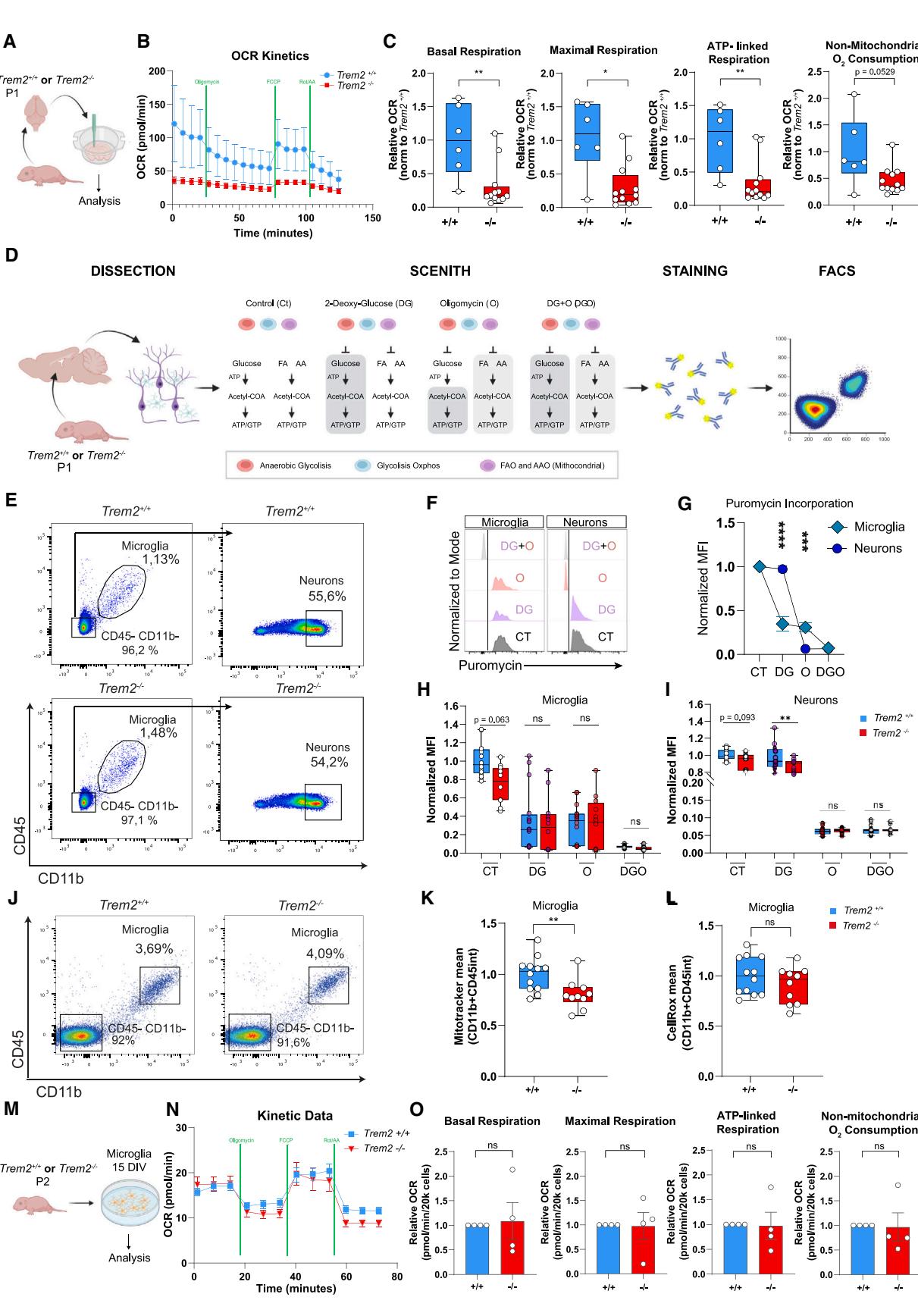
图1. 海马体新生神经元在出生后早期出现能量代谢受损 (A) 实验流程示意图 (B) 海马组织切片对不同线粒体抑制剂的平均氧消耗速率动力学响应(与新生P1幼鼠)。均值±标准误 © 从左至右:基础呼吸、最大呼吸、ATP偶联呼吸及非线粒体耗氧量。箱形图显示n=6 vs. 12个组织切片样本,来源于N=3只与新生P1幼鼠。单样本t检验,**p<0.01,*p<0.05 (D) 实验设计说明 (E) 与纯化CD11b+CD45int小胶质细胞及CD11b-CD45-NeuN+神经元的流式细胞术设门策略 (F) 代谢抑制剂处理后小胶质细胞与神经素的嘌呤霉素标记情况 (G) 小胶质细胞与神经素的嘌呤霉素平均荧光强度 (H-I) 与P1海马体小胶质细胞(H)与神经元(I)的嘌呤霉素平均荧光强度。箱形图显示N=16只、N=12只 1小鼠,含N=5次独立实验。双因素方差分析结合Tukey检验,**p<0.01 (J) 与纯化CD11b+CD45int小胶质细胞的流式设门策略 (K-L) 与P1小胶质细胞的流式线粒体追踪剂与细胞活性氧绿色染料平均荧光强度。箱形图显示N=12只、N=10只P1小鼠,含N=2次独立实验。非配对Student t检验,**p<0.01 (M) 实验流程示意图 (N) 左图:原代小胶质细胞对线粒体抑制剂的平均氧消耗速率动力学响应(与新生幼鼠) (O) 从左至右:基础呼吸、最大呼吸、ATP偶联呼吸及非线粒体耗氧量。均值±标准误,至少n=15个重复样本,来源于N=4次独立培养的与细胞。单样本t检验另见图S1
小胶质细胞可能通过Trem2塑造神经元分子图谱,而蛋白质缺失可能迫使神经元获得改变后的转录和分子图谱——这种可能性尚未被探讨。
Trem2在神经发育过程中同样发挥关键作用,它通过控制小胶质细胞介导的冗余突触清除过程,影响神经元回路形成与大脑连接性43–45。在此阶段,该蛋白表达水平较高,且随年龄增长仅出现适度提升46,47。我们聚焦于神经元成熟关键事件发生的早期发育阶段,探究Trem2是否参与神经元特性的塑造。研究发现,小胶质细胞Trem2缺失会功能性影响神经元代谢特征,这种失衡模式还与海马CA1区锥体神经元的发育轨迹延迟相关。
¶ 结果
¶ 小鼠海马体中的早期出生后神经元存在能量代谢受损
由于Trem2调控循环巨噬细胞和小胶质细胞的生物能量学,16我们通过Seahorse细胞外通量分析平台测量氧气消耗速率(OCR),研究了Trem2缺失对和小鼠P1期急性海马切片中线粒体整体代谢的影响(图1A)。OCR测量结果显示,与 (相比,出生后海马组织表现出整体线粒体呼吸动力学降低(图1B)),其中基础呼吸、最大呼吸和ATP依赖性呼吸均显著下降(图1C),表明海马组织满足整体能量需求的能力受损。P1期小鼠的海马切片还显示出较低的非线粒体相关呼吸(图1C),表明通过其他形式酶促活动氧化 的能力较低。48
我们评估了观察到的减少是否与Trem2缺失情况下的小胶质细胞改变有关。与P18阶段小鼠CA1区小胶质细胞密度低于、43的情况不同,P1阶段小鼠海马CA1或CA3区的Iba1阳性细胞数量均无显著差异(图S1A和S1C)。相反,对P1阶段 小胶质细胞的形态学分析显示,每个细胞的突起分支和连接点数量更多(图S1B、S1D和S1E),且稳态受体P2yr12的表达更高(图S1F)。这些变化仅在CA1区具有统计学意义(图1D–1F左半部分)。
为了直接区分不同细胞类型对代谢表型改变的贡献,我们采用了一种基于流式细胞术的方法,在单细胞分辨率下功能性分析能量代谢,即SCENITH(通过翻译抑制分析的单细胞能量代谢)49 (图1D)。该检测方法基于以下概念:活细胞中来自葡萄糖、氨基酸和/或脂质分解代谢的大部分能量被蛋白质合成机制消耗50。因此,蛋白质合成可作为整体代谢活性的替代测量指标。我们利用嘌呤霉素结合抗嘌呤霉素单克隆抗体49的掺入作为可靠读数,用于测量蛋白质合成51–53并在单细胞分辨率下分析代谢表型。
从P1海马体中纯化的CD11b+/CD45int小胶质细胞与NeuN+神经元均被用于实验处理(图1E)。我们发现 P1小胶质细胞在ATP供能蛋白合成过程中同等依赖糖酵解和线粒体途径,因为分别阻断糖酵解和线粒体ATP合酶的脱氧葡萄糖(DG)与寡霉素(O)处理均降低了嘌呤霉素的掺入量(图1F与1G)。与之相反,P1神经元主要依赖线粒体作为ATP来源,其在DG处理后未表现出嘌呤霉素掺入量的任何变化(图1G)。
通过比较和小胶质细胞,我们观察到其基础代谢出现轻微下降(图1H)。从P1海马体中分离的CD11b+/CD45int小胶质细胞经线粒体追踪染色后,通过对P1海马区D11b+/CD45int小胶质细胞进行流式细胞术分选后,采用Mitotracker染色进一步证实了小胶质细胞的代谢缺陷(图1J)。结果显示线粒体质量减少(图1K),但活性氧(ROS)生成未见改变(图1L)。此外,从P1 大脑分离的原代培养小胶质细胞经Seahorse能量代谢分析(图1M)显示,其代谢适应性相对于 (图1N)仅存在可忽略的损伤,主要OCR参数未出现显著变化(图1O)。
出乎意料的是,我们观察到出生后海马神经元的线粒体代谢出现显著受损(图1I),而CA1或CA3区神经元密度均未发生改变(图S1G和S1H),这表明Trem2缺失会在早期发育阶段影响神经元代谢。
¶ 马体进行单细胞测序发现,神经元中存在深度转录重组,以及代谢过程的细胞特异性参与。
为研究发育中小胶质细胞缺乏Trem2如何影响神经元行为,我们通过单细胞RNA测序(scRNA-seq)54 (对P1时期和Tre 同窝仔的海马区进行转录组分析(图2A、2B))。在两种基因型间按比例分布分析的26,326个细胞(图2C)中,无监督聚类分析识别出15个细胞簇,根据其典型特征进一步归为13种不同细胞类型(图2C与S2B)。
正如预期的那样,我们识别出神经元祖细胞(顶端祖细胞[AP]和中间祖细胞[IPC])以及胶质前体细胞(星形胶质细胞和少突胶质细胞前体)。能够区分来自内侧和尾侧神经节隆起(MGE和CGE)的CA区(CA1和CA3锥体神经元)与齿状回(颗粒细胞和苔藓细胞)的有丝分裂后兴奋性神经元不同亚类以及抑制性GABA能中间神经元。同时鉴定出一个富含未成熟CA锥体神经元的细胞簇( 细胞),凸显了该阶段神经元分化的动态特征55。我们还发现了以小胶质细胞特征性标志物Trem2、Tmem119、Iba1、Cx3cr1、Csf1r和Tyrobp表达的微胶质细胞(图S2A和S2B)。与既往报道一致,Trem2表达从出生后早期阶段46,47持续至成年期(P90,图S2C)。
通过对细胞类型进行差异丰度分析,56我们未观察到从和同窝仔中分离的P1海马体组成发生显著变化(图2C、S2D和S2E)。然而,差异基因表达分析(图2D)显示兴奋性锥体细胞和颗粒细胞的转录足迹存在显著差异(DG颗粒细胞:569个失调基因,其中279个上调、290个下调;CA1锥体细胞:487个失调基因),表明未成熟亚型(DG颗粒细胞与CA未成熟神经元)及成熟CA神经元的分子特征发生显著改变。相比之下,抑制性神经元和非神经元细胞类型(包括小胶质细胞)虽然丰度较低,但在Trem2信号缺失时仅表现出有限的转录变化(图2D)。
与我们先前在成年 小鼠中发现的区域特异性小胶质细胞缺陷相一致43,我们观察到CA-Imm、CA1-Pyr和CA3-Pyr呈现出转录失调的独特模式,且基因重叠有限(图2E)。这些结果表明,在缺乏Trem2信号传导的情况下,会触发亚型特异性反应。事实上,从敲除幼鼠脑中分离出的CA神经元亚群中,大多数表达异常的基因在各亚群中均呈现选择性改变(图2E),仅有极少数基因在其他亚群中共享表达(表S1)。所有CA神经元簇的通路富集分析(差异表达基因[DEGs])显示,各类神经元均普遍存在关键生物过程的失调(图2F;表S2),这些过程涉及翻译机制(真核翻译起始因子2[EIF2]信号通路和哺乳动物雷帕霉素靶蛋白[mTOR]通路),印证了我们此前SCENITH分析的结果(图1I)。进一步支撑区域特异性反应的是,属于mTOR和线粒体功能障碍通路的基因仅在CA1-Pyr和CA-Imm神经元中呈现特异性富集,而在CA3-Pyr中未见此现象(图S2G和S2H)。此外,代谢改变被确定为其神经元足迹的显著特征:主要失调集中于线粒体机制,且未涉及糖酵解的转录调控(图2I)。
氧化磷酸化(OXPHOS)通路在 神经元中出现严重失调。确实,我们观察到与呼吸链相关基因行为发生重要变化,这些基因编码从复合物I到V的所有复合体(复合物II除外)(图2J)。我们发现属于复合物I的基因(如Ndufa5和Ndufb2)、复合物III的基因(如Uqcrq)以及复合物IV的基因(如Cox6a1和Cox8a)在CA1锥体神经元中选择性下调,而在CA3锥体神经元中未出现此现象(图2H)。我们还观察到CA1亚型(包括CA1-Pyr和CA-Imm)特异性调控异常的复合物V基因(如Atp5e),该基因编码线粒体中负责ATP合成的ATP合酶复合体的催化单元(图2H)。
总体而言,这些证据表明,当小胶质细胞Trem2缺失时,神经元会发生深刻的转录重排,选择性地影响神经元生物能量学。与此同时,定向分化为特定区域命运(即CA1神经元)的神经细胞中,ATP合成会出现预期性抑制。
¶ 小胶质细胞TREM2缺失影响CA1神经元分化
由于线粒体代谢是大脑中细胞命运转变的关键驱动因素,57–59我们测试了在小胶质细胞Trem2缺失情况下,观察到的OXPHOS通路相关基因下调是否可能参与海马神经元群体分化/发育动态的潜在改变。为理解小胶质细胞中Trem2缺失如何影响神经元定向分化,我们通过推断轨迹拓扑结构和伪时间(图3A)来考察神经元谱系的发育路径。我们首先在海马发育标准模型中捕获所有相关细胞,并确定了对应兴奋性谱系的降维空间区域——这些细胞类型在小胶质细胞缺乏Trem2时表现出最显著的转录改变(图2D)。我们排除了抑制性中间神经元(因其前体细胞不在海马区内定位而无法捕获轨迹60)以及苔藓纤维细胞(其生成发生在海马神经发生早期阶段 E10-E12.5)。61
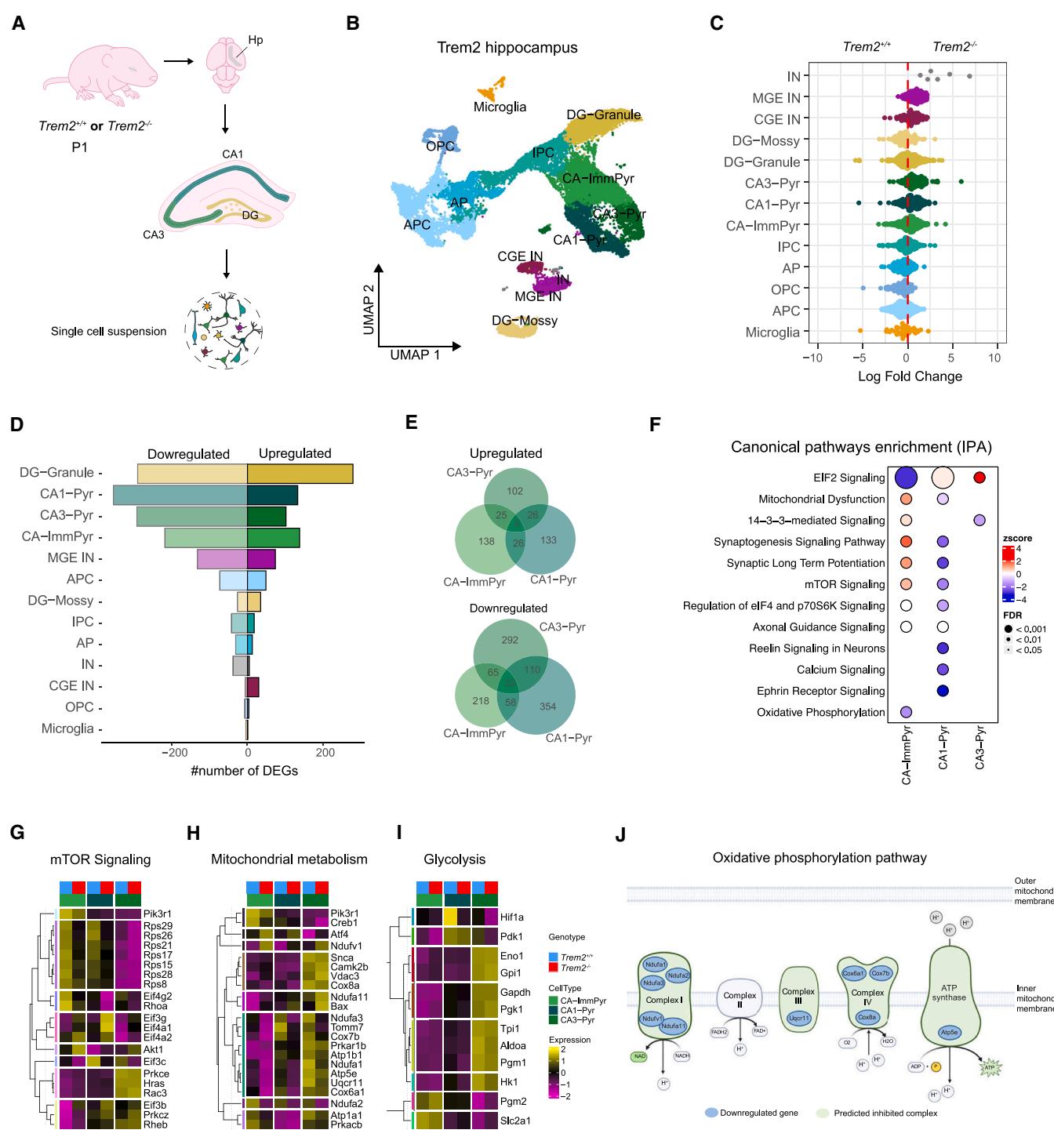
图2. 与基因P1海马组织的单细胞转录组分析揭示发育中神经元的代谢重编程
(A) P1 和小鼠海马组织分离与解离示意图。(B) 按细胞类型标注着色的均匀流形逼近与投影图。© 不同基因型间细胞丰度变化的蜂群图。(D)各细胞类型差异表达基因数量的条形图(单细胞转录组模型基础分析[MAST],校正后p值 )。(E) 显示选定细胞类型间差异表达基因重叠的维恩图。(F) 各细胞类型通路富集分析点图。(G–I) 选定细胞类型的平均表达热图,显示与特定通路相关的失调基因。(J) 预测的抑制复合物及OXPHOS通路中下调基因示意图。另见图S2。
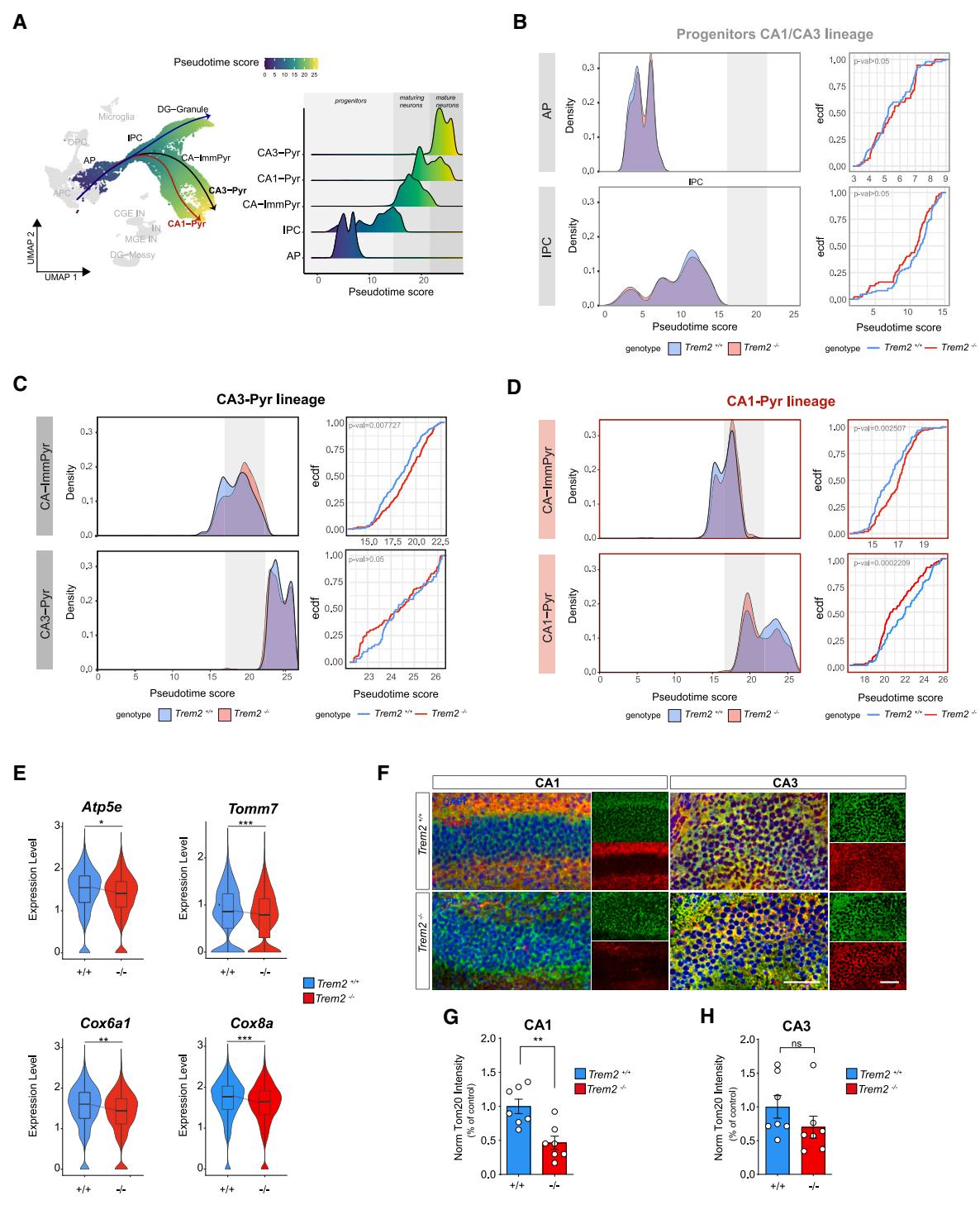
图3. 新生小鼠线粒体缺陷特异性破坏CA1神经元的成熟(A) 显示谱系轨迹的UMAP图。按细胞类型着色的拟时序分值密度图。 (B–D) 密度图和经验累积分布函数图显示各谱系中不同基因型细胞沿拟时序的分布情况(KS检验)。 (E) 在拟时序区间= 17-22内、按基因型分组的线粒体功能相关差异表达基因小提琴图。MAST-re统计检验,*p adj < 0.05,**p adj < 0.01,***p adj < 0.001。 (F) 与海马体P1时期CA1区(上图)和CA3区(下图)代表性共聚焦图像,采用双皮质素(Dcx)(绿色)和Tom20(红色)抗体标记。比例尺75 mm。 (G和H) 分别为CA1和CA3锥体层中Tom20平均荧光强度。数据表示为N= 7只与 P1-2幼崽的平均值± SEM。数据经对应值标准化。非配对Student t检验,**p< 0.01。另见图S3。
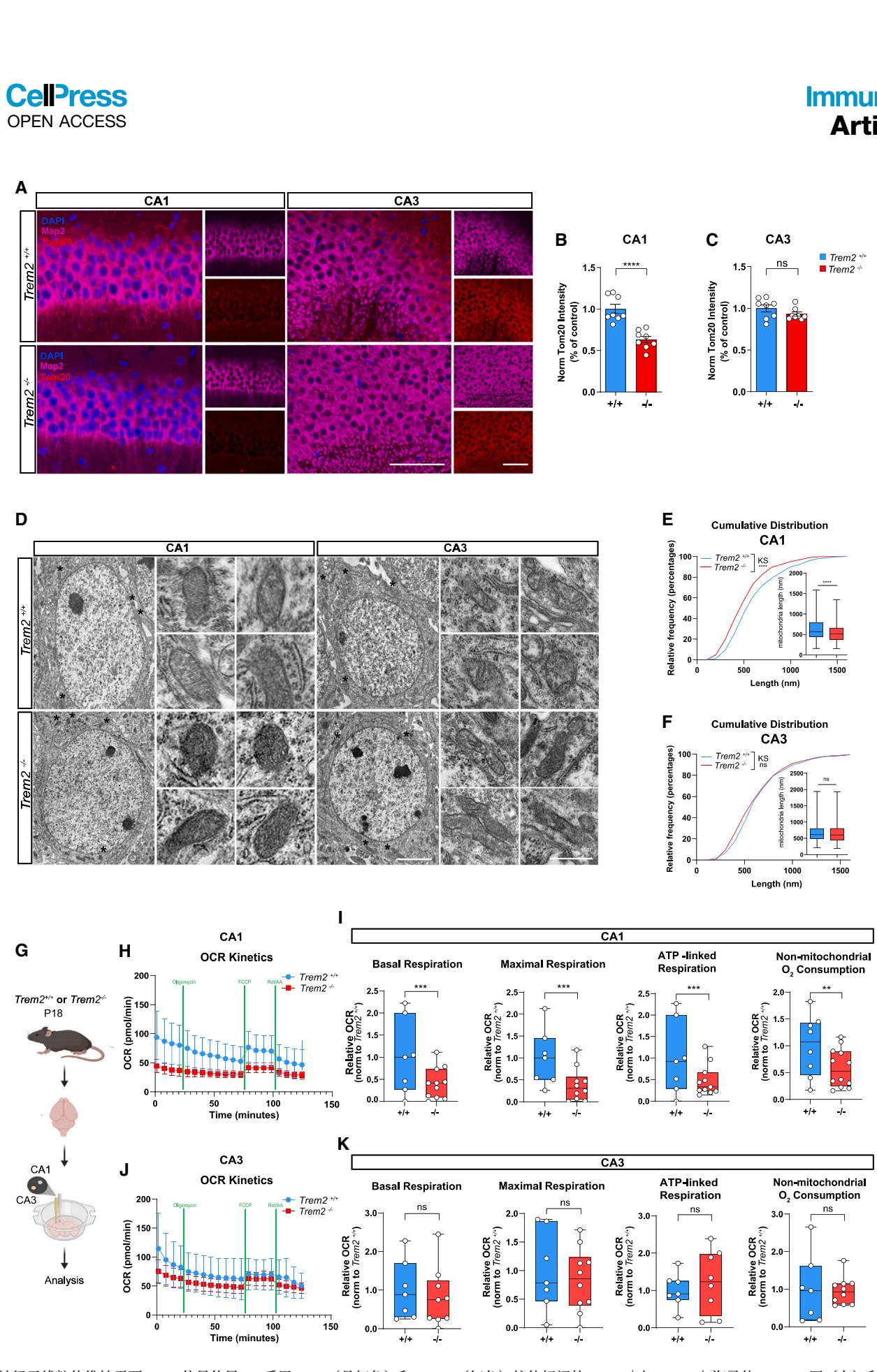
图4. 神经元线粒体维持需要Trem2信号传导�(A) 采用Map2(品红色)和Tom20(红色)抗体标记的与海马体P18 CA1区(左)和CA3区(右)亚域的共聚焦图像。比例尺标度为75毫米。(B和C) 分别为CA1区(B)和CA3区©锥体细胞层中Tom20平均强度的定量分析。数据来自N=8只和的P18小鼠,表示为均值±标准误。非配对Student t检验,****p < 0.0001。
(D) P18 和小鼠CA1区(左)和CA3区(右)锥体神经元胞体的拼图扫描电子显微照片。比例尺:3微米。星号指示胞体线粒体,右侧显示了其放大图。比例尺:650纳米。
(E和F) CA1和CA3区胞体中线粒体长度的累积分布曲线及箱型图。CA1区KS检验p < 0.0001,CA3区KS检验p > 0.05。箱型图数据来自N=3只和 P18小鼠的>300个线粒体。非配对Student t检验,****p < 0.0001。
(G) 实验流程示意图。
(H) 平均氧消耗速率动力学图,显示来自和 P18小鼠CA1亚区脑片的打孔样本对不同线粒体抑制剂的反应。均值±标准误。
(I) 从左至右:CA1脑片打孔样本的基础呼吸、最大呼吸、ATP耦联呼吸及非线粒体氧耗。箱型图数据来自N=3只和 P18小鼠,分别为n=8和n=12个样本。单样本t检验,*p < 0.001及p < 0.01。
(J) 平均氧消耗速率动力学图,显示来自和 P18小鼠CA3亚区脑片的打孔样本对不同线粒体抑制剂的反应。均值±标准误。
(K) 从左至右:CA3脑片打孔样本的基础呼吸、最大呼吸、ATP耦联呼吸及非线粒体氧耗。箱型图数据来自N=3只和 P18小鼠,分别为n=7和n=10个样本。单样本t检验,p < 0.001,p < 0.01及p < 0.05。
另见图S4。
在轨迹推断过程中,我们检测到由多种细胞类型代表的三个不同兴奋性谱系(图3A)。从AP的共同起点(“根”)出发,轨迹通过IPC进行并分裂成三个不同的亚系,代表三种不同神经元亚型的发育命运获取,这些亚系由不同的末端分支定义(图3A)。在 和对照组同窝仔之间未观察到谱系拓扑结构的变化;然而,伪时序分析揭示了不同条件下沿轨迹分支的细胞分布差异(图3B-3D)。密度分布分析(图S3A和S3B)显示沿伪时序的细胞频率存在显著差异,特别是CA1谱系(Kolmogorov-Smirnov [KS]检验: ),而另外两个细胞谱系未见显著差异(KS: )(图S3A和S3B)。
我们还通过解析两种基因型的单个细胞类型对每个谱系进行了分析,重点关注CA神经元。分析结果显示,CA1和CA3谱系的共同祖细胞(AP和IPC)沿轨迹呈现相似分布(图3B和图S3C)。对CA-ImmPyr密度分布的分析显示,在中间伪时序区间(伪时序评分17-22)内,两个谱系均出现显著增加(CA3谱系,KS: 0.007727,图3C上图;CA1谱系,KS: ,图3D上图),该区间还包含来自Trem 的成熟CA1神经元(与相比)。值得注意的是,仅在CA1-Pyr神经元细胞类型中观察到中间伪时序区间的细胞积累伴随终末分化状态(伪时序评分22-25)的显著减少(KS: ,图3C下图),而CA3-Pyr神经元中未出现此现象(KS: ,图3D下图)。这些数据为Trem2缺失情况下兴奋性神经元发育动力学受损提供了支持,并特别强调了影响兴奋性CA1神经元的成熟延迟现象。
接下来,我们研究了在特定伪时间值区间(17-22,灰色区域)内表现出不同密度的细胞之间基因表达的变化,并在条件间显著失调的基因中鉴定出关键线粒体基因:线粒体外膜的结构调节因子(如Tomm7)对线粒体复合物的组装至关重要,在细胞中显示出显著下调,同时线粒体呼吸机制的关键功能调节因子(包括Atp5e、Cox6a1、Cox7c、Cox7b、Cox7a2、Cox8a和Cox20)也出现下调(图3E;表S1)(校正后p值 )。
最后,为评估海马神经元中观察到的线粒体通路转录组失调是否反映了线粒体结构的改变,我们对和 小鼠脑组织的P1切片进行了外膜转位酶复合体亚基20(Tom20,图3F)免疫染色——这是线粒体质量的可靠标记物62。共聚焦分析显示,与 (相比,海马体的Tom20平均强度降低,且仅在CA1区域达到显著水平(图3G和3H))。
总体而言,这些数据表明小胶质细胞Trem2的缺失会诱导锥体神经元发育轨迹的改变,其中CA1谱系出现特异性延迟效应,该现象与调控线粒体组装的基因程序改变相关。
¶ 神经元中的线粒体维持需要TREM2信号传导
线粒体是动态细胞器,通过不断分裂与融合重塑形态,以整合细胞的生物能量学与信号传导功能63。在海马体的CA1区(而非CA3区)检测到Tom20平均强度降低,这一现象甚至出现在发育后期(P18天,图4A-4C)。通过透射电子显微镜(TEM)对P18天与海马体CA1、CA3区线粒体结构的定量分析(图4D)显示,CA1区(而非CA3区)锥体神经元胞体中的线粒体平均长度显著缩短(图4E、4F)。
与线粒体超微结构反映其功能状态相一致,64 对P18周龄和小鼠CA1、CA3区域组织穿刺样本的OCR测量(图4G)显示:CA1亚区(图4H和4I)在所有OCR参数上均存在选择性损伤,但CA3亚区(图4J和4K)则未出现此现象。
这些发现表明,小胶质细胞中Trem2的遗传性缺失会引发CA1靶向的氧化磷酸化链选择性损伤,并伴随线粒体结构改变,且这种损伤在发育后期阶段仍持续存在。
¶ CA1海马区正常发育需要高水平的TREM2表达
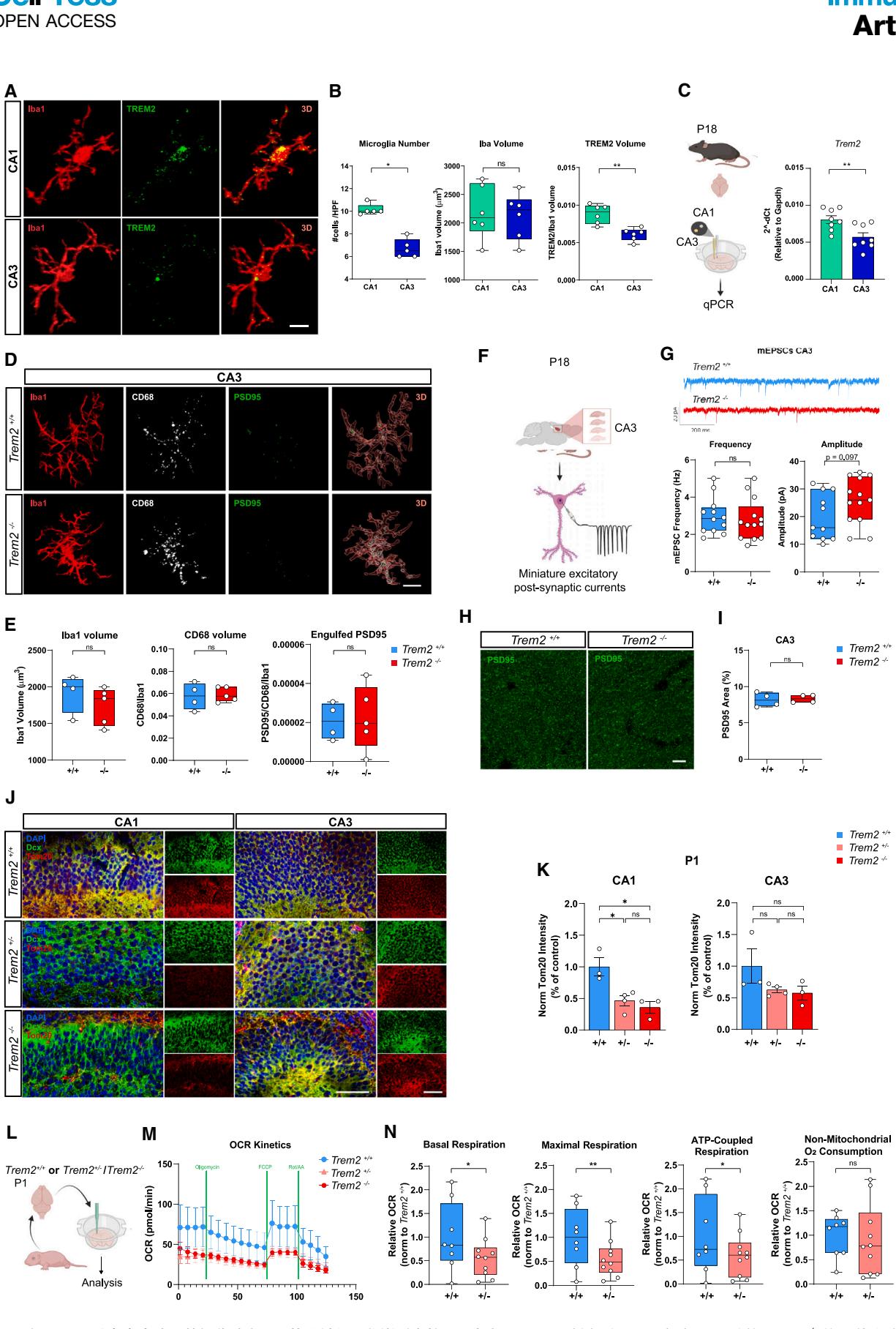
图5. Trem2 在 CA1 区域高度表达,其部分减少足以扰乱神经元代谢适应性 (A) 来自 P18 CA1(上)和 CA3(下)子区域的 海马体小胶质细胞的共聚焦图像和 3D 重建,用抗 Iba1(红色)和 Trem2(绿色)的抗体标记。比例尺,10 毫米。
接下来,我们探究了Trem2缺失在CA1锥体神经元中产生更显著效应的基础机制。如先前所示⁴³,P18阶段的小胶质细胞在CA1区域的富集程度高于CA3区域(图5A和5B左侧面板),而Iba1染色显示其细胞体积无显著差异(图5A和5B中间图)。共聚焦定量分析表明,Trem2相对于Iba1的表达量在CA1区显著高于CA3区(图5A、5B右图)。与此一致的是,对P18海马切片不同区域进行1毫米穿刺实时定量PCR检测,发现CA1区Trem2 mRNA含量更高(图5C)。此外,虽然P18 小鼠海马CA1区小胶质细胞对冗余突触的吞噬作用受损,但我们在CA3区未检测到突触吞噬(图5D、5E)、微小兴奋性突触后电流频率(mEPSCs,图5F、5G)或PSD95标记的兴奋性突触密度(图5H、5I)存在显著差异。因此与CA1区不同,CA3区受Trem2基因缺失的影响较小,表明该海马区域可能存在独立于Trem2的不同作用机制。对体感皮层(SSCx)的分析显示,其小胶质细胞密度显著低于海马CA1区(图S5B),且单细胞Trem2表达量略低(图S5C左图)。全视野Trem2体积定量证实CA1区蛋白表达显著更高,而CA3区与SSCx无差异(图S5C右图)。此外与CA3区类似,与小鼠在SSCx的Tom20表达未见差异(图S5E)。由此可见,海马CA1区功能的正常发挥似乎更显著依赖于携带Trem2的小胶质细胞。
为评估CA1区域是否严格依赖高Trem2表达的小胶质细胞,我们利用 /(半合子)小鼠验证:即使Trem2表达部分下调,是否足以诱发神经元代谢紊乱。我们发现,无论是出生后第1天(图5J和5K)还是出生后第18天(图S5F)的海马体,其Tom20平均强度均出现降低,尤其在CA1区域表现显著。因此,OCR测量显示P1 海马体,类似于 (图1A–1C),显示总体降低的线粒体呼吸动力学(图5M和5N),表明它们满足整体能量需求的能力受损。
这些数据表明,CA1海马区域的神经元需要高水平的微胶质细胞Trem2以进行适当的分化和功能,并且即使Trem2的部分减少就足以扰乱神经元的代谢适应性。
¶ 来自小鼠的CA1海马神经元显示突触改变
我们接下来探究 P18海马神经元中的代谢改变是否会影响突触功能和神经传递。缺乏Trem2的P18小鼠在CA1区表现出显著增多的兴奋性突触,这源于突触修剪期间小胶质细胞对冗余突触清除功能的缺失43。此处我们分析了这些突触的超微结构特征。通过透射电镜观察发现, 小鼠CA1锥体层的突触中线粒体长度显著缩短(图6A和S6A),同时伴随突触面积减小和突触小泡密度增加(图6B和S6B),但活性区长度和锚定突触小泡密度未见差异(图6B和S6B)。与CA1突触不同,CA3区突触在线粒体长度、突触面积或突触小泡密度方面均未显示改变(图6C、6D、S6C和S6D),活性区长度和锚定小泡密度亦无变化(图6D和S6D)。
为了探究缺乏小胶质细胞Trem2的P18小鼠体内兼具CA1和CA3区域的神经元网络功能,我们采用多电极阵列电生理学技术,该技术可同步获取神经元群体场电位电活动的高通量读数(图6E和图S6E)。我们将平均放电率作为自发活动的表征指标进行测量在整个海马切片(图6E)中观察神经元活动,发现 P18切片CA1区(图6F)的多单元放电频率显著升高,而CA3区(图6G)未见此现象。该效应主要源于神经元自发性活动的平均水平增强,而非放电细胞数量增加65 ((图S6F))。与组相比, 海马体还表现出更高的同步性与连接性(图6H与6I)。这种自发性神经元活动的增强与我们早先在P18 小鼠中验证的兴奋性突触标记物密度升高及微型事件增多现象相吻合43。
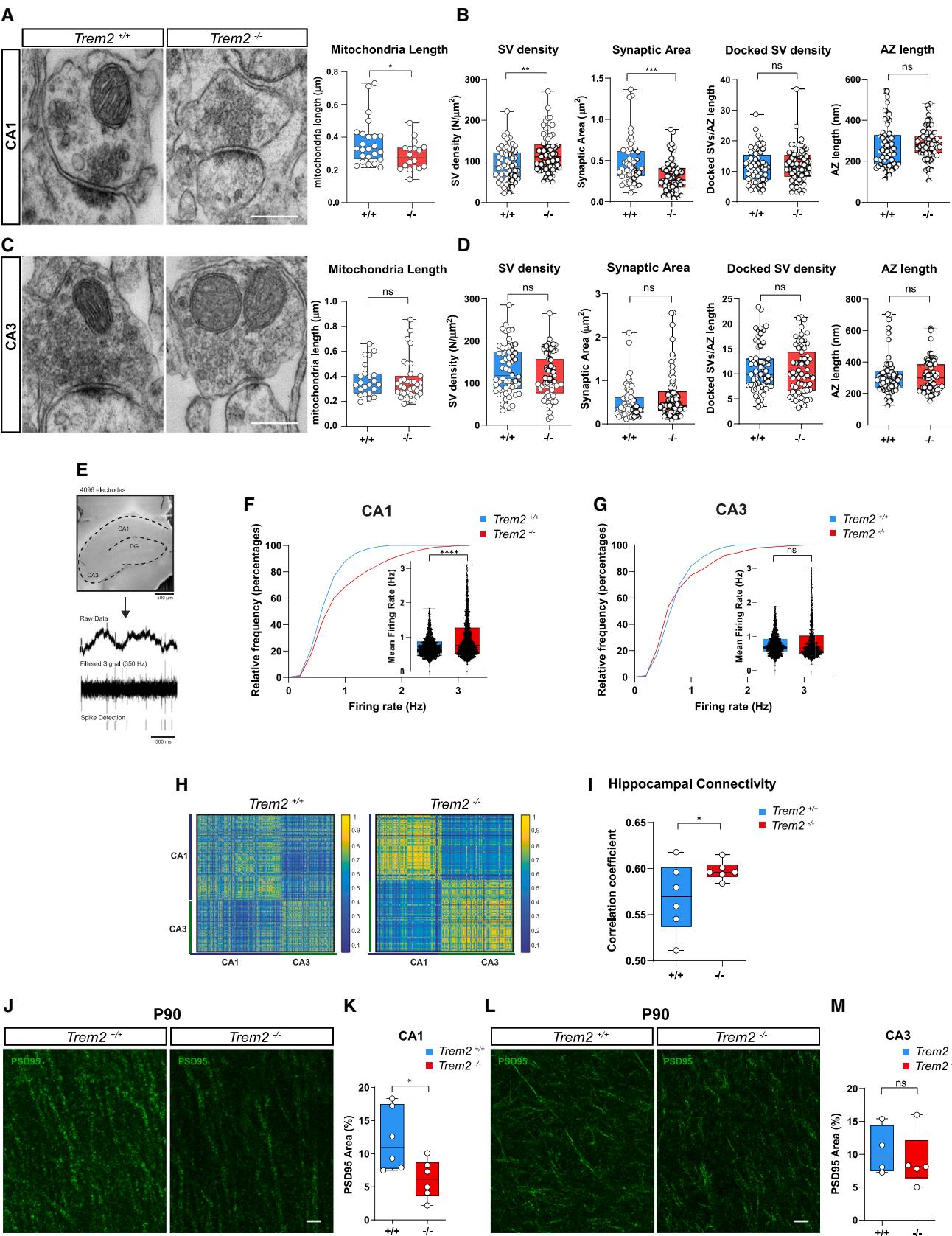
图6. 海马神经元以区域依赖性方式呈现突触和功能改变。(A) P18 与小鼠CA1锥体层突触的电子显微图像。比例尺为200纳米。箱线图显示来自 只与 P18小鼠的 个( )或18个( )突触的线粒体长度。曼-惠特尼U检验, 。
(B) P18 和 小鼠 CA1 区锥体层突触的突触囊泡密度、突触面积、锚定突触囊泡密度以及活动区长度。箱形图数据来源于 N=3 只 和 小鼠的 n=56 () 或 59个突触。Mann-Whitney U 检验,**p < 0.01 及 ***p < 0.001。
© 左图:P18 和 小鼠 CA3 区锥体层突触的电子显微照片。比例尺:200 纳米。右图:突触内线粒体长度的箱形图,数据来源于 N=3 只 和 P18 小鼠的 n=23 () 或 35 () 个突触。Mann-Whitney U 检验。
(D) 和 小鼠 CA3 区锥体层突触的突触囊泡密度、突触面积、锚定突触囊泡密度以及活动区长度。箱形图数据来源于 N=3 只 和 小鼠的 n=58 () 或 63 () 个突触。Mann-Whitney U 检验。
(E) 上图:P18 小鼠海马冠状切片在 CMOS 多电极阵列上的图像。比例尺:500 微米。下图:源自一个电极的原始数据、滤波信号(350 Hz)及锋电位检测。
(F 和 G) 和 P18 小鼠 CA1 区 (F) 和 CA3 区 (G) 神经元平均自发放电频率的累积频率分布及定量分析。箱形图数据来源于 3 只小鼠的 N=6 个海马切片中记录的 n>600 个神经元。点代表单个放电单元。非配对 Student t 检验,****p < 0.0001。
(H) 和 P18 雄性小鼠在 CA1 和 CA3 区自发活动期间神经元对的互相关矩阵。
(I) 和 小鼠 CA1 和 CA3 区域内所有活动神经元对的平均相关系数。箱形图数据来源于 N=3 只 P18 和 小鼠的 n=6 个切片。单侧非配对 Student t 检验,*p < 0.05。
(J 和 K) 用抗 PSD95 抗体标记的 P90 和 小鼠海马 CA1 区 (J) 和 CA3 区 (K) 的共聚焦图像。比例尺:10 微米。
(L 和 M) CA1 区 (L) 和 CA3 区 (M) 的 PSD95 体积分数。箱形图数据来源于 N=6 只 和 N=6 只 P90 小鼠。非配对 Student t 检验。
另见图 S6。
我们先前已证实,在更成熟的发育阶段(P90),与P18时期相比,海马体中PSD95蛋白含量出现下降,但树突棘密度未发生改变43。通过特异性分析不同海马区域,本研究进一步发现小鼠在P90时期出现的突触退化现象特异性发生于CA1区,而CA3区未见异常(图6J-6M)。这些数据表明, CA1区的代谢缺陷可能导致成年期(P90)突触强化功能受损。
¶ 来自小鼠的神经元即使在与大脑环境分离后,仍表现出线粒体缺陷和分化异常。
我们接下来想知道, 大脑神经元中检测到的代谢紊乱是否需要小胶质细胞的持续邻近作用,还是依赖于细胞自主机制。为探究这一问题,我们从和小鼠P1期海马体中提取原代神经元,在体外培养4天(4 DIV)后进行OCR测定(图7A)。结果显示神经元的线粒体功能完整性发生显著改变(图7B)。实际上,培养4天的 神经元表现出能量需求满足能力受损(图7C)。 神经元的非线粒体呼吸功能也存在缺陷(图7C)。基于单细胞RNA测序显现的转录特征——这些特征涉及大多数与OXPHOS和线粒体功能障碍通路相关的基因——这些数据表明代谢功能完整性的缺陷在早期发育阶段就已通过转录机制确立,且这一特征从根本上预先决定了线粒体功能结果方面,为与4 DIV培养物中细胞外通量测量观察到的神经元代谢紊乱相呼应,在神经元的胞体和神经突中均检测到Tom20显著减少(图7D),与 (相比(图7E和7F))。此外,通过活体成像实验在神经元胞体处检测到线粒体ATP产量下降(图7G)。同时,Sholl分析(图7H)显示神经元整体分支化程度(图7I)和初级神经突长度(图7J)均有缩减,这支持了神经元发育成熟进程延迟的假说。
鉴于Na+和K+通道的组成随着神经元细胞的成熟会发生显著变化66,我们对培养4天的神经元进行了电生理学表征。通过记录电流-电压(I-V)关系来评估内向Na+电流和外向K+电流(图7K)。I-V曲线显示,与 神经元相比, 神经元的 电流密度显著降低,而 电流密度无显著变化(图7L和7M)。此外, 神经元的静息膜电位( )较神经元更趋于去极化(图7N),这通常被认为是神经元未成熟的特征67,68。最后, 神经元相较于 (神经元表现出略高的电阻率( )和更低的电容值( )(图7O))。这些数据表明,即使在原代培养条件下,来自 海马的神经元也表现出代谢特性缺陷和成熟延迟,这与在体实验观察结果相吻合(图1A-1C、3A-3D和3F-3H)。
我们接下来要探究的是,在P18阶段体内观察到的兴奋性神经传递紊乱(图6E-6I)是否同样存在于 海马成熟培养神经元中。我们前期已证明,来自或小鼠的14天体外培养神经元电生理特性并无差异43。在P18脑片的CA1和CA3亚区(图S6G、S6H)以及14天体外培养神经元中(图S7D、S7E),两组神经元的静息电位与被动特性(电阻率;电容)也表现相似。然而与离体实验情况类似,在小鼠的14天体外培养神经元胞体与突触接触点中,线粒体功能缺陷依然存在(图 S5A–S5C)。此外,在14 DIV 突触的突触终扣处进行活体成像记录,使用谷氨酸感应荧光报告剂(iGluSNFR)显示,与对照组培养相比,谷氨酸释放显著增加(图7P和7Q)。为评估这些突触对线粒体ATP的依赖性,我们在同一突触处记录了高频刺激下线粒体ATP的波动情况,并使用ATP红色染料检测突触ATP波动,发现突触无法响应高频刺激产生ATP(图7R),这与线粒体功能降低的结论一致。
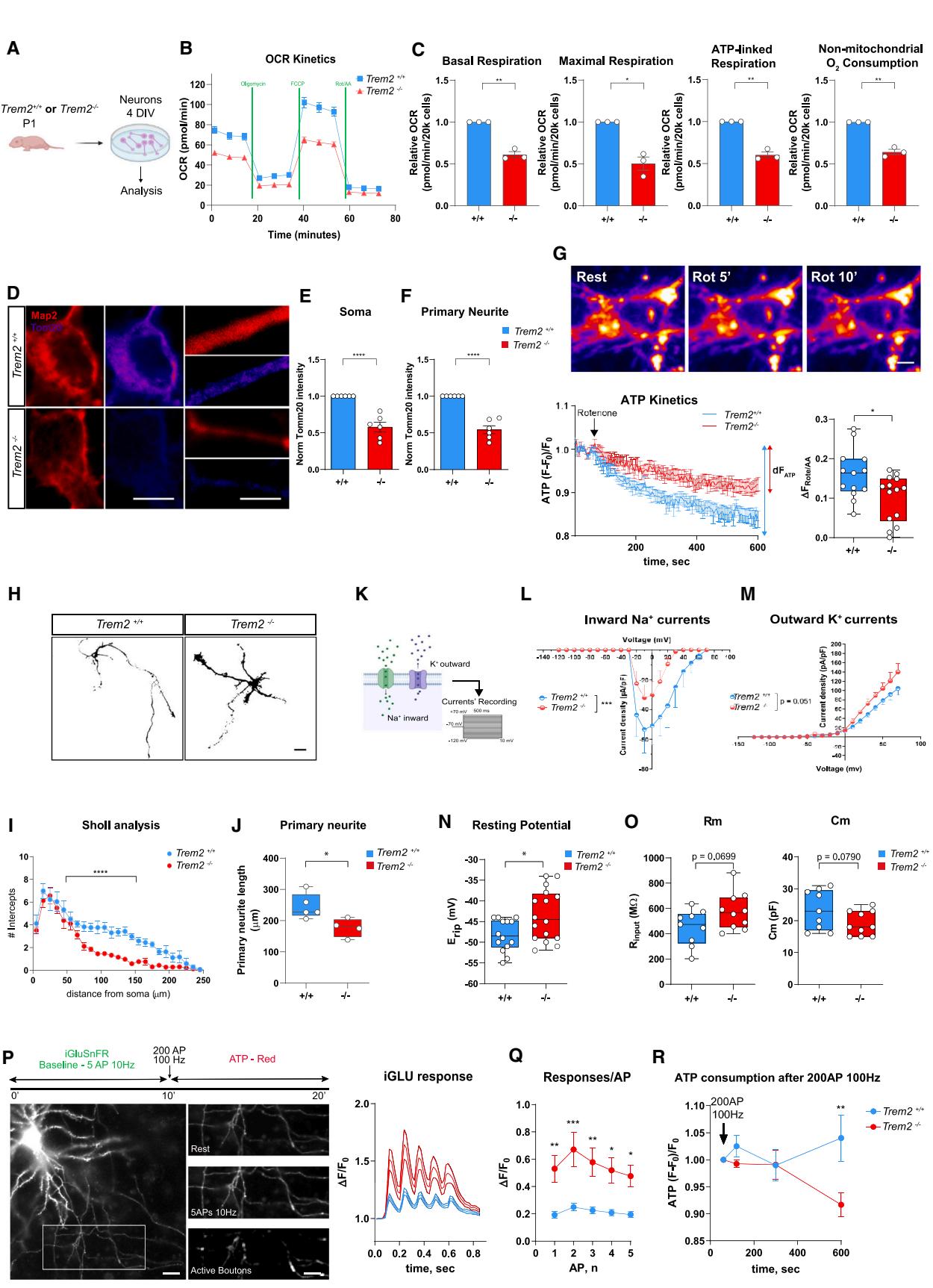
图7. 神经元在培养中表现出线粒体缺陷和分化异常 (A) 实验设计示意图 (B) 平均OCR动力学曲线显示培养4天的或T 原代海马神经元对不同线粒体抑制剂的反应。
© 从左至右:基础、最大、ATP耦联及非线粒体氧消耗速率。数据来自 N=3 个独立的 和 培养体系,n=25 个重复样本,表示为均值±标准误。单样本 t 检验,****p < 0.0001。
(D) 用抗 Tom20 抗体(伪彩色)和抗 MAP2 抗体(红色)标记的 4 DIV 或 原代海马神经元的共聚焦图像。比例尺:8 微米和 4 微米。
(E 和 F) 分别为胞体 (E) 和初级神经突 (F) 的 Tom20 平均强度。数据来自 N=6 个独立的 和 神经元培养体系,n=40 () 和 n=40 () 个神经元,表示为均值±标准误。非配对 t 检验,****p < 0.0001。
(G) 上图:在 4 DIV 神经元胞体中,药物抑制线粒体复合物 1 和 3(鱼藤酮/抗霉素A)后,代表性 ATP 红色荧光强度的变化。比例尺:2 微米。下图左:胞体处的平均 ATP 红色荧光强度变化曲线 (ΔF/F0)。数据表示为均值±标准误。下图右:处理后 15 分钟 ATP 减少程度 (ΔFRote/AA) 的统计图。数据来自 N=4 个 和 神经元培养体系,n=13 () 和 n=14 () 个神经元的箱型图。非配对 Student t 检验,*p < 0.05。
(H) 用抗 MAP2 抗体标记并进行 Sholl 分析处理的 4 DIV 或 原代海马神经元的共聚焦图像。比例尺:20 微米。
(I) 4 DIV 或 原代海马神经元的平均树突分支情况。数据来自 N=5 个独立的 或 N=4 个 原代神经元培养体系,n=54 () 和 n=41 () 个神经元,表示为均值±标准误。双因素方差分析及 Sidak 多重比较检验,****p < 0.0001。
(J) 4 DIV 或 原代海马神经元的初级神经突长度。数据来自 N=5 个独立的 或 N=4 个 原代神经元培养体系,n=54 () 和 41 () 个神经元的箱型图。非配对 t 检验,*p < 0.05。
(K) 实验设计示意图。
(L 和 M) 4 DIV 或 原代海马神经元的平均内向 Na+ 电流和外向 K+ 电流。数据来自 N=3 个 或 原代神经元培养体系,n=5–10 () 和 n=6–9 () 个神经元。双因素方差分析,***p < 0.001。
(N) 4 DIV 或 原代海马神经元的静息电位。数据来自 N=4 个 或 原代神经元培养体系,n=14 () 和 n=16 () 个神经元的箱型图。Mann-Whitney U 检验,*p < 0.05。
(O) 4 DIV 或 原代海马神经元的电阻值(左)和电容值(右)。数据来自 N=3 个 和 神经元培养体系,n=9 () 和 n=10 () 个神经元的箱型图。非配对 t 检验。
§ 上图:iGluSNFR 活细胞成像实验设计描述。下图:转染的 神经元中代表性的 iGluSNFR 荧光图像。右侧为给予 5 个动作电位(10 Hz)刺激后,带有活动性突触终扣的轴突分支放大图像。比例尺:20 微米和 5 微米。左图: 和 突触对 10 Hz 频率下 5 个动作电位的 iGluSNFR 反应平均曲线。数据来自来源于 P1 或 幼鼠的 N=5–6 个海马培养体系,n=22 () 或 n=21 () 个盖玻片,表示为均值±标准误。
(Q) ΔF/F0 反应随动作电位数量变化的统计图。双因素方差分析及 Sidak 多重比较检验,p < 0.05,p < 0.01,p < 0.001。
® 或 突触在 100 Hz 频率下 200 个动作电位刺激后 ATP 红色荧光强度变化过程的平均曲线。双因素方差分析及 Sidak 多重比较检验,**p < 0.01。
为了开始评估在缺乏小胶质细胞Trem2的情况下发育的神经元的缺陷代谢表型是否可能源于早期神经元暴露于改变的小胶质细胞分泌组,69–72 从或原代小胶质细胞收集的预条件培养基(CM)被应用于从或 海马体分离的原代神经元,并维持4天体外培养(图S7F)。Seahorse实验(图S7G)显示,与神经元相比, 神经元的代谢参数存在缺陷。然而,释放的可溶性因子并未增强任何代谢参数(图S7H–S7K),上清液的效果非常微弱,不显著,无论其是来源于还是小胶质细胞。
综上所述,这些结果表明,在早期发育关键期小胶质细胞与神经元之间缺乏Trem2介导的通讯,足以扰乱后续神经元代谢和线粒体组织结构。这种异常状态甚至在神经元脱离大脑环境后被依然保持。这些缺陷与 神经元成熟延迟相关,并在体外实验中并且在体外,通过谷氨酸能神经传递失调。此外, 小胶质细胞的分泌组无法逆转神经元缺陷的代谢谱,这表明可能需要更结构化的双向小胶质细胞-神经元通讯。
¶ 讨论
小胶质细胞是高度动态的细胞,它们在大脑实质中巡逻,感知神经元活动,并通过释放多种免疫调节因子来调节神经元功能。通过分泌性及细胞间接触介导的过程,小胶质细胞动态调控神经元的发育、生长、突触形成以及脑细胞的生理功能。1,73 我们先前已证实,免疫受体Trem2的缺失会导致小胶质细胞在大脑发育期间无法正常执行冗余突触清除功能。43 此外,我们发现成年期(P90)Trem2缺陷小鼠表现出脑连接性改变和行为缺陷。43,45 本研究进一步证明,Trem2缺失会损害神经元转录组学和能量代谢特征,表明神经元的早期代谢紊乱可能导致成年Trem2缺陷小鼠/出现连接性和行为缺陷。
Trem2的缺失强烈影响神经元中EIF2信号的表达,该信号在CA-ImmPyr中显著下调,而在CA1-Pyr和CA3-Pyr中上调。通过控制eIF2·GTP·Met-tRNAiMet三元复合物,EIF2活性主导全局翻译速率,其磷酸化作用被证实与神经系统发育及记忆巩固相关。74,75事实上,基因层面的Elf2b活性会诱导延迟的产后大脑发育、异常的胶质细胞丰度、脱髓鞘轴突数量增加,以及在受到损伤时大脑炎症反应受损。76,77
本研究通过多条证据表明,在小胶质细胞Trem2缺失的情况下,CA1神经元的线粒体功能与能量代谢系统会发生深刻改变。小胶质细胞能够通过免疫代谢过程灵活重编程其代谢途径,从而应对环境变化。78,79 事实上,稳态小胶质细胞主要依赖氧化磷酸化产生ATP,而炎症状态下的小胶质细胞会将代谢模式转向有氧糖酵解。80–82 此外,Trem2在调节细胞生物合成代谢中起关键作用16,并核心参与诱导转录组和功能程序向吞噬态小胶质细胞(DAMs)转化——这是在神经退行性疾病大脑中发现的一种吞噬状态26,33。
我们发现,在出生后的早期阶段,小胶质细胞同样依赖糖酵解和氧化磷酸化来供应ATP。我们还表明,与Trem2在控制小胶质细胞代谢中的核心作用一致,16该基因的缺失在出生时会导致线粒体质量减少,但并未引起OCR的显著变化。相反,来自/小鼠的海马神经元表现出线粒体代谢受损,基础呼吸、最大呼吸和ATP耦合呼吸选择性恶化,同时伴有线粒体质量减少和线粒体长度缩短。因此,小胶质细胞Trem2的缺失主要影响神经元的代谢特性。
与小胶质细胞不同,神经元仅具有有限程度的代谢灵活性,这是确保命运稳定性和维持细胞功能所必需的。在发育过程中,干细胞和前体细胞持续的代谢性糖酵解重编程调控着细胞命运决定、增殖与分化进程。相反,成熟神经元为优先维持网络功能,主要依赖氧化磷酸化和线粒体供能,仅有极少部分能量来源于负责调控突触小泡运输等过程的糖酵解途径8384。有研究提出,糖酵解或某些糖酵解代谢物会与神经元身份维持及存活相冲突83,导致神经细胞去分化、细胞命运稳定性丧失、突触传递缺陷,最终引发细胞死亡。与从糖酵解向氧化磷酸化的代谢转换代表神经元成熟关键步骤的观点一致,氧化磷酸化相关基因缺陷会促使神经发育障碍的发生85,86。在小鼠中观察到的神经元氧化磷酸化水平降低,很可能阻碍了正常神经元与突触的成熟进程,甚至可能影响脑环路发育。
我们发现, 小鼠海马CA1区锥体神经元的延迟成熟现象,在出生后第18天会随之出现兴奋性神经传递的增强。类似的现象在其他神经发育障碍中也有报道,例如Rett综合征、唐氏综合征以及癫痫性脑病。这些疾病中,大脑能量功能障碍与神经元发育迟缓相关,并伴随兴奋性传递增强87,88,这种现象与神经网络同步性提升存在关联65,89。此外,糖酵解(而非线粒体)产生的ATP被用于突触小泡中谷氨酸的聚集过程90。这些数据为我们观察到的现象提供了合理解释:存在缺陷的神经元线粒体呼吸在受到刺激时会释放更大量的谷氨酸。在大脑发育的后期阶段(P90), 小鼠表现出海马突触标志物的减少43,这种现象特异地发生在CA1区。该事件与功能连接性改变及神经环路成熟过程中的显著缺陷相关联43,45。因此小鼠似乎无法在大脑发育期间正常构建和强化正确的突触结构,这可能是由于神经元代谢障碍以及突触修剪期间突触消除功能缺陷共同导致的结果。
近期大量证据表明,线粒体在早期大脑发育中起着重要作用。线粒体功能与神经发生、神经元分化及成熟密切相关。在皮质发生中期,线粒体的组装、融合/分裂及运动动力学是维持祖细胞自我更新能力的关键过程57–59。融合/分裂模式的时间性改变会影响神经发生91,而线粒体运动能力和能量代谢的缺陷会选择性阻碍皮质中间神经元的神经元分化与迁移92。此外,线粒体生物发生对海马神经元树突棘的形成与维持至关重要93。据我们所知,目前仅发现细胞自主性机制在支撑神经元代谢程序中的作用。本研究揭示了小胶质细胞在调控神经元成熟与代谢适应性方面的功能,并确定Trem2信号传导是这种小胶质细胞-神经元交互作用的早期潜在调节因子。
除DG神经元外,我们检测到特定类别神经元存在选择性易损性,并将CA1神经元确定为对Trem2缺失最敏感的神经亚型之一。与此一致的是,线粒体缺陷和代谢紊乱特异性地发生在CA1区域而非CA3区域,且在后期(P18)发育阶段更为严重。通过伪时序分析对新生小鼠海马神经元发育轨迹的评估同样显示,CA1神经元的代谢特征发生明显转变,并伴随神经元成熟延迟。我们前期研究已表明,与CA3区域相比,CA1区域的突触修剪更主要依赖于Trem2。43 因此,Trem2的多种功能是以区域特异性方式实现的。虽然CA1区域对Trem2缺失具有选择性易损性的基础尚未完全明确,但值得注意的是CA1小胶质细胞表达更高水平的Trem2。此外,即使是Trem2表达的部分降低(如Trem2杂合子小鼠),也足以扰乱CA1锥体神经元代谢,表明CA1区域需要高水平的小胶质细胞Trem2才能正确调控锥体神经元分化。这些发现具有重要意义,提示在Trem2表达降低的情况下(包括患者携带的Trem2杂合错义变异34,38,94),可能会引发神经元代谢功能障碍。
Trem2如何调节小胶质细胞-神经元通讯仍有待阐明。一种可能性是Trem2缺失及随之引发的小胶质细胞状态改变,可能导致分泌组改变,进而差异性影响神经元发育轨迹。另一种可能是,早期发育阶段形成的小胶质细胞-神经元接触可能发挥作用——这种机制也可能与前者并存。具体而言,定位于小胶质细胞膜的Trem2可与未成熟神经元对应物结合,从而影响其代谢轨迹。研究发现,在整个胚胎期和成年期神经发生过程中,小胶质细胞突起可与发育中神经元的胞体形成特异性接触,其特征是功能性小胶质细胞P2yr12受体簇集并富含线粒体。95 缺乏小胶质细胞P2yr12会导致皮层细胞结构紊乱,并持续至成年期。95,96 这种对兴奋性细胞的选择性影响(这些细胞本地产生于海马体,所有发育阶段都可能与具有指导作用的小胶质细胞接触)符合细胞间接触依赖机制的可能性。我们的伪时间分析表明,这种发生在神经元兴奋性谱系成熟早期的推定相互作用,可能产生潜在的"启动"效应,且在Trem2表达程度不同的CA1与CA3区域可能具有差异。
目前,我们尚不清楚小胶质细胞是仅在发育阶段调控神经元代谢成熟,还是在不同时间窗口中都持续发挥作用。衰老过程中神经元葡萄糖代谢会发生改变,当衰老神经元出现线粒体功能障碍时,会增强通过糖酵解途径氧化葡萄糖的能力97。衰老期间亦会出现Trem2表达下调98,这表明这两种机制可能共同促使衰老期神经元代谢从氧化磷酸化向糖酵解转换。实际上,通过氟代脱氧葡萄糖微型正电子发射断层扫描(FDG-mPET)分析显示,Trem 小鼠脑部葡萄糖代谢水平降低99;而疾病相关Trem2 T66M突变敲入小鼠模型则表现出小胶质细胞活性和全脑葡萄糖代谢的显著下降38。尽管PET检测的总FDG摄取量是神经元、星形胶质细胞和小胶质细胞代谢特征的综合体现,但类似于早期发育阶段小胶质细胞与神经元之间的交互作用,这种细胞通讯在衰老过程中可能同样存在,一旦受损便会推动神经退行性进程。在这方面特别值得注意的是,Trem2缺失的功能性后果之一是会引发谷氨酸释放增加和神经元自发性活动增强。多年研究已知海马体是阿尔茨海默病(AD)的重要受累脑区100,且在斑块形成前CA1区就会出现过度活跃神经元的增多101,102。与对照组相比,轻度AD患者的CA1区顶端神经纤维网会出现变薄103,而晚期该区域则呈现更严重的突触改变104。重要的是,本研究发现成年(P90)Trem 小鼠存在显著的突触 impoverishment,这种变化特异性地发生于CA1区,并与先前报道的连接组学和行为缺陷共存43,45。因此,存在这样一种可能:小胶质细胞Trem2功能缺陷导致对神经元代谢的调控失常,进而引起衰老和神经退行性病变期间的神经元活动紊乱——这一机制值得深入探究。
¶ 究的局限性
虽然揭示了TREM2在早期神经发育中的新作用,但本研究仍未能阐明携带Trem2的小胶质细胞调控代谢神经元命运的分子机制。需要开展更深入的分析,探究可溶性Trem2或小胶质细胞来源的细胞外囊泡105的潜在作用,并解析调控此过程的分子编码小胶质细胞与神经元之间的通讯,包括CA1和CA3区域是否差异表达推定的Trem2相互作用蛋白。此外,在出生后早期窗口期进行小胶质细胞Trem2表达下调的实验,将明确证明成年小鼠中观察到的突触和神经环路缺陷源于早期发育阶段小胶质细胞-神经元串扰的失调。更重要的是,由于Trem2缺失会同时损害突触精细化43和神经元代谢(本研究),未来需要进一步界定这两个过程在大脑成熟期间对突触形成的相对贡献程度。
¶ 星 方法
本文在线版本提供了详细方法,包括以下内容:
- 主要资源表
- 资源可用性
- 主要联系人
- 材料可用性
- 数据与代码可用性
- 方法细节
- 实验小鼠
- 原代海马神经元培养与转染
- 原代小胶质细胞培养
- 活细胞成像实验
- RNA测序分析
- 电生理学
- 免疫荧光分析
- 电子显微镜技术
¶ 补充信息
补充信息可在 https://doi.org/10.1016/j.immuni.2023.12.002 上在线查阅。
¶ 致谢
我们感谢K. Volynski教授(实验与临床癫痫学系)惠赠iGluSnFR和GFP质粒。感谢C. Ferrari、D. Pozzi、E. Fraviga、C.A. Elia和C. Saulle在建立实验方法学中提供的帮助。本研究受ERC AdG MATILDA 101055323、EraNET Neuron JTC2021 InflASD及Telethon GGP20030资助(授予M.M.),ERC StG IMPACT 101043003和Cariplo Giovani 2019-1785资助(授予S.L.),以及重型设备拨款D.R. 3404(日立120kV透射电镜HT7800,授予K.C.)。E.T.和A.C.获Humanitas研究基金会高级博士后(HiPPO)奖学金支持。插图和图形摘要通过Biorender.com制作。
¶ 作者贡献
E.T.负责原代神经元和小胶质细胞培养,开展并分析体外实时成像实验、离体电子显微镜研究以及体外和离体免疫荧光实验,同时协助样本采集。G.D.负责原代神经元培养,开展并分析体外和离体Seahorse实验,并协助样本收集。M.B.负责原代小胶质细胞培养、免疫荧光实验、条件培养基制备及样本采集。E.F.负责qPCR、菌株维持、基因分型与样本收集。A.C.、R.J.A.和G.D.共同设计SCENITH实验,并由A.C.完成实验操作。F.F.与A.C.共同建立并完成FACS实验。S.M.、M.M.和S.L.
执行、分析并解读了单细胞测序数据。M.C.G.和K.C.进行了离体电子显微镜实验及数据分析。R.M.开展了体外电生理记录、免疫荧光实验,并协助样本制备。R.H.-S.完成MEA记录,P.P.执行了离体免疫荧光实验。M.M.、E.T.、G.D.和R.M.共同构思本研究、设计实验方案并进行数据分析。M.M.、E.T.、G.D.和S.L.撰写了论文初稿,所有作者均参与了稿件修订。
¶ 利益声明
作者声明无竞争性利益。
收到日期:2022年9月15日
修订日期:2023年7月17日
接受日期:2023年12月5日
发表日期:2023年12月29日
¶ 参考文献
- Colonna, M. 与 Butovsky, O. (2017). 小胶质细胞在健康与神经退行性病变期间于中枢神经系统中的功能。免疫学年评 35, 441–468.
- Hammond, T.R., Dufort, C., Dissing-Olesen, L., Giera, S., Young, A., Wysoker, A., Walker, A.J., Gergits,F., Segel, M., Nemesh, J. 等 (2019). 小鼠全生命周期及脑损伤状态下小胶质细胞的单细胞RNA测序揭示复杂细胞状态变化。免疫 50, 253–271.e6.
- Hughes, A.N. 与 Appel, B. (2020). 小胶质细胞通过吞噬髓鞘调节发育期髓鞘形成。自然神经科学 23, 1055–1066.
- Paolicelli, R.C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., Giustetto, M., Ferreira, T.A., Guiducci, E., Dumas, L. 等 (2011). 小胶质细胞介导的突触修剪对正常脑发育至关重要。科学 333, 1456–1458.
- Paolicelli, R.C., Sierra, A., Stevens, B., Tremblay, M.E., Aguzzi, A., Ajami, B.,Amit, I., Audinat, E., Bechmann, I., Bennett, M. 等 (2022). 小胶质细胞状态与命名法:处于十字路口的研究领域。神经元 110, 3458–3483.
- Schafer, D.P., Lehrman, E.K., Kautzman, A.G., Koyama, R., Mardinly, A.R., Yamasaki, R., Ransohoff, R.M., Greenberg, M.E., Barres, B.A., Stevens, B. (2012). 小胶质细胞通过活动性与补体依赖方式塑造出生后神经环路。神经元 74, 691–705.
- Sierra, A., Encinas, J.M., Deudero, J.J.P., Chancey, J.H., Enikolopov, G., Overstreet-Wadiche, L.S., Tsirka, S.E., Maletic-Savatic, M. (2010). 小胶质细胞通过凋亡耦联的吞噬作用调控成年海马神经发生。细胞干细胞 7, 483–495.
- Thion, M.S., Ginhoux, F., Garel, S. (2018). 小胶质细胞与早期脑发育:一段密切相伴的旅程。科学 362, 185–189.
- Zhan, Y., Paolicelli, R.C., Sforazzini, F., Weinhard, L., Bolasco, G., Pagani, F., Vyssotski, A.L., Bifone, A., Gozzi, A., Ragozzino, D. 等 (2014). 神经元-小胶质细胞信号传导缺陷导致脑功能连接与社会行为受损。自然神经科学 17, 400–406.
- Colonna, M. (2023). TREM受体的生物学特性。自然免疫学评论 23, 580–594.
- Kiialainen, A., Hovanes, K., Paloneva, J., Kopra, O., Peltonen, L. (2005). 参与先天免疫与神经退行性病变的Dap12和Trem2在中枢神经系统中共表达。神经生物学疾病 18, 314–322.
- Neumann, H., Takahashi, K. (2007). 髓系细胞表达触发受体-2(TREM2)对中枢神经组织免疫稳态的重要作用。神经免疫学杂志 184, 92–99.
- Schmid, C.D., Sautkulis, L.N., Danielson, P.E., Cooper, J., Hasel, K.W., Hilbush, B.S., Sutcliffe, J.G., Carson, M.J. (2002). 成年小鼠小胶质细胞中髓系细胞触发受体-2的异质性表达。神经化学杂志 83, 1309–1320.
- 高桥康弘、罗克福德·C·D·P与诺伊曼·H (2005)。小胶质细胞髓系细胞触发受体-2介导的神经元凋亡清除而不引发炎症。《实验医学杂志》201, 647–657。
- 特恩布尔·I·R、吉尔菲兰·S、切拉·M、青石俊司、米勒·M、皮乔·L、埃尔南德斯·M与科隆纳·M (2006)。研究前沿:TREM-2可减弱巨噬细胞活化。《免疫学杂志》177, 3520–3524。
- 乌兰·T·K、宋·W·M、黄·S·C-C、乌尔里希·J·D、谢尔古什切夫·A、比蒂·W·L、洛博达·A·A、周·Y、凯恩斯·N·J、坎巴尔·A等 (2017)。TREM-2维持阿尔茨海默病中小胶质细胞的代谢适应性。《细胞》170, 649–663.e13。
- 菲利佩洛·F、游·S·F、米尔法哈尔·F·S、马哈利·S、博尔曼·B、阿夸罗内·M、科尔瓦茨卡·O、马什·J·A、西瓦拉曼·A、马丁内斯·R等 (2023)。携带TREM2功能缺失突变的人小胶质细胞在溶酶体功能与脂质代谢中的缺陷。《神经病理学学报》145, 749–772。
- 纽金特、A.A.、林、K.、范伦格里奇、B.、利亚诺格洛、S.、普日比拉、L.、戴维斯、S.S.、拉帕什蒂卡、C.、王、J.、金、D.J.、夏、D.等(2020)。TREM2在长期吞噬挑战下调节小胶质细胞胆固醇代谢。《神经元》105卷,837–854.e9。
- van Lengerich, B., Zhan, L., Xia, D., Chan, D., Joy, D., Park, J.I., Tatarakis, D.,Calvert, M., Hummel, S., Lianoglou, S., 等. (2023). 一种具有血脑屏障转运载体的TREM2激活抗体可增强阿尔茨海默病模型中的小胶质细胞代谢。 Nat. Neurosci. 26, 416–429.
- 王, S., 苏丹, R., 彭, V., 周, Y., 杜, S., Yuede, C.M., 雷, T., 侯, J., 蔡, Z., 塞拉,M., 等. (2022). TREM2通过SYK依赖性与非依赖性通路驱动小胶质细胞对β-淀粉样蛋白的反应。《细胞》185卷, 4153–4169.e19页.
- Cignarella, F., Filipello, F., Bollman, B., Cantoni, C., Locca, A., Mikesell, R.,Manis, M., Ibrahim, A., Deng, L., Benitez, B.A., 等 (2020). 小胶质细胞上的TREM2激活促进多发性硬化症模型中的髓鞘碎片清除和髓鞘再生。《神经病理学学报》140, 513–534.
- Kober, D.L. 与 Brett, T.J. (2017). 健康与疾病中的TREM2-配体相互作用. 《分子生物学杂志》429, 1607–1629.
- 奥特罗, K., 筱原, M., 赵, H., 切拉, M., 吉尔菲兰, S., 科卢奇, A., 法乔, R., 罗斯, F.P., 泰特尔鲍姆, S.L., 高柳, H., 等. (2012). TREM2和β-连环蛋白通过控制破骨细胞生成速率调节骨稳态. 《免疫学杂志》第188卷,第2612–2621页.
- 王莹、Cella, M.、Mallinson, K.、Ulrich, J.D.、Young, K.L.、Robinette, M.L.、Gilfillan, S.、Krishnan, G.M.、Sudhakar, S.、Zinselmeyer, B.H.等(2015)。TREM2脂质感知维持阿尔茨海默病模型中的小胶质细胞反应。《细胞》杂志160卷,1061–1071页。
- Yeh, F.L.、Wang, Y.、Tom, I.、Gonzalez, L.C.和Sheng, M. (2016). TREM2通过与载脂蛋白(包括APOE和CLU/APOJ)结合,从而促进小胶质细胞对β-淀粉样蛋白的摄取。Neuron 91, 328–340.
- 凯伦-肖尔, H., 斯宾拉德, A., 韦纳, A., 马特科维奇-纳坦, O., 德维尔-斯特恩菲尔德, R., 乌兰, T.K., 戴维, E., 巴鲁克, K., 拉拉-阿斯塔伊索, D., 托特, B., 等. (2017). 一种与限制阿尔茨海默病发展相关的独特小胶质细胞类型. Cell 169, 1276–1290.e17.
- 费尔巴希, D., 辛德勒, P., 巴尔斯克, C., 约勒, S., 本-卢卡, E., 沃林格, K.A.,科米内尼, S., 凯卡斯, A., 何, D.J., 叶, C., 等 (2017). ADAM17是生成人髓系细胞表达触发受体(hTREM2)胞外域的主要脱落酶,并在组氨酸157位点切割TREM2。神经科学快报 660, 109–114。
- Wunderlich, P., Glebov, K., Kemmerling, N., Tien, N.T., Neumann, H., 与 Walter, J. (2013). 髓系细胞表达触发受体-2(TREM2)通过胞外域脱落和γ-分泌酶依赖性膜内切割的连续蛋白水解过程。《生物化学杂志》288, 33027–33036.
- Del-Aguila, J.L., Benitez, B.A., Li, Z., Dube, U., Mihindukulasuriya, K.A., Budde, J.P., Farias, F.H.G., Ferna´ndez, M.V., Ibanez, L., Jiang, S., 等. (2019). TREM2在阿尔茨海默病及TREM2突变携带者中的脑转录本特异性研究. 分子神经退行性病变 14, 18.
- Filipello, F., Goldsbury, C., You, S.F., Locca, A., Karch, C.M., and Piccio, L. (2 022). 可溶性TREM2:神经系统疾病的无辜旁观者还是积极参与者?《神经生 物学疾病》165, 105630.
- Piccio, L., Buonsanti, C., Cella, M., Tassi, I., Schmidt, R.E., Fenoglio, C., Rinker, J., Naismith, R.T., Panina-Bordignon, P., Passini, N., 等 (2008). 脑脊液中可溶性TREM-2的鉴定及其与多发性硬化症和中枢神经系统炎 症的关联。《大脑》131, 3081–3091.
- Sua´ rez-Calvet, M., Kleinberger, G., Ara que Caballero, M.A´ ., Brendel, M., Rominger, A., Alcolea, D., Fortea, J., Lleo´ , A., Blesa, R., Gispert, J.D., 等 (2016). sTREM2脑脊液水平是早期阿尔茨海默病小胶 质细胞活性的潜在生物标志物并与神经元损伤标志物相关。《EMBO分子医 学》8, 466–476.
- Deczkowska, A., Keren-Shaul, H., Weiner, A., Colonna, M., S chwartz, M., and Amit, I. (2018). 疾病相关小胶质细胞:神经退行性变的通用免 疫传感器。《细胞》173, 1073–1081.
- Guerreiro, R., Wojtas, A., Bras, J., Carra squillo, M., Rogaeva, E., Majounie, E., Cruchaga, C., Sassi, C., Kauwe, J.S.K., You nkin, S., 等 (2013). 阿尔茨海默病中的TREM2变异体。《新英格兰医学杂志》3 68, 117–127.
- Jin, S.C., Benitez, B.A., Karch, C.M., Cooper, B., Skorupa, T., Car rell, D., Norton, J.B., Hsu, S., Harari, O., Cai, Y., 等 (2014). TREM2编码变异增加 阿尔茨海默病风险。《人类分子遗传学》23, 5838–5846.
- Jonsson, T., Stefans son, H., Steinberg, S., Jonsdottir, I., Jonsson, P.V., Snaedal, J., Bjornsson, S., Hutte nlocher, J., Levey, A.I., Lah, J.J., 等 (2013). 与阿尔茨海默病风险相关的TREM2 变异体。《新英格兰医学杂志》368, 107–116.
- Yeh, F.L., Hansen, D.V., and Sheng, M. (2017). TREM2、小胶质细胞与神经退行性疾病。《分子医学趋势 》23, 512–533.
- Kleinberger, G., Yamanishi, Y., Sua´ rez-Calvet, M., Czirr, E., L ohmann, E., Cuyvers, E., Struyfs, H., Pettkus, N., Wenninger-Weinzierl, A., Mazah eri, F., 等 (2014). 神经退行性疾病相关的TREM2突变损害细胞表面运输和吞噬 功能。《科学转化医学》6, 243ra86.
- Krasemann, S., Madore, C., Cialic, R., B aufeld, C., Calcagno, N., El Fatimy, R., Beckers, L., O’Loughlin, E., Xu, Y., Fane k, Z., 等 (2017). TREM2-APOE通路驱动神经退行性疾病中功能障碍小胶质细 胞的转录表型。《免疫力》47, 566–581.e9.
- Yuan, P., Condello, C., Keene, C. D., Wang, Y., Bird, T.D., Paul, S.M., Luo, W., Colonna, M., Baddeley, D., and Grut zendler, J. (2016). 小鼠和人类TREM2单倍体不足会损害小胶质细胞屏障功能导 致淀粉样蛋白压缩减少和严重轴突营养不良。《神经元》90, 724–739.
- Cant oni, C., Bollman, B., Licastro, D., Xie, M., Mikesell, R., Schmidt, R., Yuede, C.M., Galimberti, D., Olivecrona, G., Klein, R.S., 等 (2015). TREM2在体内调节小胶质 细胞对脱髓鞘反应的激活。《神经病理学学报》129, 429–447.
- Dong, Y., D ’Mello, C., Pinsky, W., Lozinski, B.M., Kaushik, D.K., Ghorbani, S., Moezzi, D., Brown, D., Melo, F.C., Zandee, S., 等 (2021). 在多发性硬化症病灶中发现的氧化 磷脂酰胆碱介导神经变性并被小胶质细胞中和。《自然神经科学》24, 489–50 3.
- Filipello, F., Morini, R., Corradini, I., Zerbi, V., Canzi, A., Michalski, B., Erre ni, M., Markicevic, M., Starvaggi-Cucuzza, C., Otero, K., 等 (2018). 小胶质细胞先 天免疫受体TREM2是突触消除和正常脑连接所必需的。《免疫力》48, 979–9 91.e8.
- Scott-Hewitt, N., Perrucci, F., Morini, R., Erreni, M., Mahoney, M., Witk owska, A., Carey, A., Faggiani, E., Schuetz, L.T., Mason, S., 等 (2020). 磷脂酰丝 氨酸的局部外化介导小胶质细胞发育性突触修剪。《EMBO杂志》39, e10538 0.
- Zerbi, V., Pagani, M., Markicevic, M., Matteoli, M., Pozzi, D., Fagiolini, M., Bozzi, Y., Galbusera, A., Scattoni, M.L., Provenzano, G., 等 (2021). 对16种自闭症小鼠模型进行脑图谱测绘揭示功能连接亚型谱系。Mol. Psy chiatry 26, 7610–7620.
- Chertoff, M., Shrivastava, K., Gonzalez, B., Acarin, L. 与 Gime´nez-Llort, L. (2013). 小鼠产后脑发育过程中TREM2蛋白的差异调节. P LoS One 8, e72083.
- Thrash, J.C., Torbett, B.E. 与 Carson, M.J. (2009). TREM2 与DAP12在小鼠中枢神经系统的发育调控:对Nasu-Hakola病的启示. Neuroche m. Res. 34, 38–45.
- Herst, P.M., Tan, A.S., Scarlett, D.J. 与 Berridge, M.V. (200 4). 线粒体基因敲除细胞的细胞表面耗氧量. Biochim. Biophys. Acta 1656, 79–87 .
- Arg€uello, R.J., Combes, A.J., Char, R., Gigan, J.P., Baaziz, A.I., Bousiquot, E ., Camosseto, V., Samad, B., Tsui, J., Yan, P. 等 (2020). SCENITH:基于流式细 胞术的单细胞分辨率能量代谢功能分析方法. Cell Metab. 32, 1063–1075.e7.
- Buttgereit, F. 与 Brand, M.D. (1995). 哺乳动物细胞中ATP消耗过程的层级结构. Biochem. J. 312, 163–167.
- Aviner, R. (2020). 嘌呤霉素的科学:从核糖体功 能研究到生物技术应用. Comput. Struct. Biotechnol. J. 18, 1074–1083.
- Ranga raju, V., Lauterbach, M. 与 Schuman, E.M. (2019). 空间稳定的线粒体区室在可塑 性期间驱动局部翻译. Cell 176, 73–84.e15.
- Hidalgo San Jose, L. 与 Signer, R. A.J. (2019). 体内蛋白质合成的细胞类型特异性定量. Nat. Protoc. 14, 441–460.
- Mancinelli, S. 与 Lodato, S. (2018). 解码发育中大脑皮层的神经元多样性:从 单细胞到功能网络. Curr. Opin. Neurobiol. 53, 146–155.
- La Manno, G., Soldat ov, R., Zeisel, A., Braun, E., Hochgerner, H., Petukhov, V., Lidschreiber, K., Kastri ti, M.E., Lo¨nnerberg, P., Furlan, A. 等 (2018). 单细胞RNA速率. Nature 560, 494– 498.
- Dann, E., Henderson, N.C., Teichmann, S.A., Morgan, M.D. 与 Marioni, J. C. (2022). 基于k近邻图的单细胞数据差异丰度检验. Nat. Biotechnol. 40, 245–25 3.
- Brunetti, D., Dykstra, W., Le, S., Zink, A. 与 Prigione, A. (2021). 线粒体在 神经发生中的作用:对线粒体疾病的启示. Stem Cells 39, 1289–1297.
- Iwata, R., Casimir, P., Erkol, E., Boubakar, L., Planque, M., Gallego Lo´pez, I.M., Ditkows ka, M., Gaspariunaite, V., Beckers, S., Remans, D. 等 (2023). 线粒体代谢设定神 经元发育的物种特异性节律. Science 379, eabn4705.
- Knobloch, M. 与 Jessber ger, S. (2017). 代谢与神经发生. Curr. Opin. Neurobiol. 42, 45–52.
- Asgarian, Z ., Magno, L., Ktena, N., Harris, K.D. 与 Kessaris, N. (2019). 海马CA1区生长抑素 中间神经元起源于胚胎期MGE/POA. Stem Cell Rep. 13, 793–802.
- Save, L., Baude, A. 与 Cossart, R. (2019). 胚胎起源时间关键决定齿状回细胞生理特性. C ereb. Cortex 29, 2639–2652.
- Roche, M.E., Lin, Z., Whitaker-Menezes, D., Zhan , T., Szuhai, K., Bovee, J.V.M.G., Abraham, J.A., Jiang, W., Martinez-Outschoorn, U. 与 Basu-Mallick, A. (2020). 线粒体外膜转位酶复合体亚基20(TOMM20) 促进软骨肉瘤侵袭性与治疗抵抗. Biochim. Biophys. Acta Mol. Basis Dis. 1866, 165962.
- Heine, K.B. 与 Hood, W.R. (2020). 线粒体行为、形态与动物表现. B iol. Rev. Camb. Philos. Soc. 95, 730–737.
- Chen, H. 与 Chan, D.C. (2009). 神经 退行性疾病中的线粒体动力学——融合、分裂、移动及线粒体自噬. Hum. Mol. Genet. 18, R169–R176.
- Stampanoni Bassi, M., Iezzi, E., Gilio, L., Centonze, D., and Buttari, F. (2019).突触可塑性塑造大脑连接:对网络拓扑结构的启示。《国际分子科学杂志》20.
- Song, M., Mohamad, O., Chen, D., and Yu, S.P. (2013). 电压门控Na+与K+电流的协同发育调控人诱导多能干细胞来源前脑神经元的功能成熟。《干细胞与发育》22卷, 1551–1563.
- Luhmann, H.J., Reiprich, R.A., Hanganu, I., andKilb, W. (2000). 新生大鼠大脑皮层的细胞生理学:内在膜特性与钠钙电流。《神经科学研究杂志》62卷, 574–584.
- Spruston, N., and Johnston, D. (1992).海马三类神经元被动膜特性的穿孔膜片钳分析。《神经生理学杂志》67卷, 508–529.
- Basso, M., and Bonetto, V. (2016). 细胞外囊泡与大脑中新型通信形式。《神经科学前沿》10卷, 127.
- Gabrielli, M., Raffaele, S., Fumagalli, M., and Verderio, C. (2022). 小胶质细胞释放细胞外囊泡的多重面孔:十年研究进展回顾?《细胞神经科学前沿》16卷, 984690.
- Paolicelli, R.C., Bergamini, G., and Rajendran, L. (2019). 细胞外囊泡的细胞间通信:聚焦小胶质细胞。《神经科学》405卷, 148–157.
- Rufino-Ramos, D., Albuquerque, P.R., Carmona, V., Perfeito, R., Nobre, R.J., and Pereira de Almeida, L. (2017). 细胞外囊泡:脑部疾病治疗的新型递送系统。《控制释放杂志》262卷, 247–258.
- Szepesi, Z., Manouchehrian, O., Bachiller, S., and Deierborg, T. (2018). 健康与疾病状态下小胶质细胞-神经元的双向通信。《细胞神经科学前沿》12卷, 323.
- Pavitt, G.D. (2005). eIF2B:通用及基因特异性翻译控制的介导因子。《生物化学学会汇刊》33卷, 1487–1492.
- Bellato, H.M., and Hajj, G.N.M. (2016). eIF2α在神经元中的翻译控制:超越应激反应。《细胞骨架》73卷, 551–565.
- Cabilly, Y., Barbi, M., Geva, M., Marom, L., Chetrit, D., Ehrlich, M., and Elroy-Stein, O. (2012). eIF2B敲入小鼠脑炎症反应缺陷:对白质消融病病因学的启示。《PLoS综合》7卷, e46715.
- Fogli, A., and Boespflug-Tanguy, O. (2006). eIF2B相关疾病的大谱系。《生物化学学会汇刊》34卷, 22–29.
- Hotamisligil, G.S. (2017). 免疫代谢的基础及其对代谢健康与疾病的意义。《免疫》47卷, 406–420.
- Makowski, L., Chaib, M., and Rathmell, J.C. (2020). 免疫代谢:从基础机制到转化应用。《免疫学评论》295卷, 5–14.
- Cheng, J., Zhang, R., Xu, Z., Ke, Y., Sun, R.,Yang, H., Zhang, X., Zhen, X., and Zheng, L.T. (2021). 早期糖酵解重编程调控小胶质细胞炎症活化。《神经炎症杂志》18卷, 129.
- Holland, R., McIntosh, A.L., Finucane, O.M., Mela, V., Rubio-Araiz, A., Timmons, G., McCarthy, S.A., Gun’ko, Y.K., and Lynch, M.A. (2018). 炎症小胶质细胞呈现糖酵解与铁滞留特征并代表APP/PS1小鼠模型中的小胶质细胞类型。《大脑行为与免疫》68卷, 183–196.
- Lauro, C., and Limatola, C. (2020). 小胶质细胞代谢重编程在先天炎症反应调控中的作用。《免疫学前沿》11卷, 493.
- Traxler, L., Lagerwall, J., Eichhorner, S., Stefanoni, D., D’Alessandro, A., and Mertens, J. (2021). 代谢调控发育、衰老与神经退行中的神经细胞命运。《疾病模型与机制》14卷, 8993.
- Ashrafi, G., Wu, Z., Farrell, R.J., and Ryan, T.A. (2017). GLUT4动员支持活性突触的能量需求。《神经元》93卷, 606–615.e3.
- Janssen, R.J., van den Heuvel, L.P., and Smeitink, J.A. (2004). 氧化磷酸化系统的遗传缺陷。《分子诊断专家评论》4卷, 143–156.
- Rojas-Charry, L., Nardi, L., Methner, A., and Schmeisser, M.J. (2021). 自闭症谱系障碍及相关神经发育障碍中突触线粒体异常。《分子医学杂志》(柏林)99, 161–178.
- McGrath, T., Baskerville, R., Rogero, M., and Castell, L. (2022). 谷氨酸能功能障碍在神经精神疾病中广泛作用的新证据。《营养素》14, 917.
- Oyarza´bal, A., Musokhranova, U., Barros, L., and Garcı´a-Cazorla, A. (2021). 儿童神经发育障碍中的能量代谢。EBiomedicine 69, 103474.
- Li, X., and Ascoli, G.A.(2008). 突触同步对神经元输入输出关系的影响。《神经计算》20, 1717–1731.
- Ikemoto, A., Bole, D.G., and Ueda, T. (2003). 突触小泡中的糖酵解和谷氨酸积累。甘油醛磷酸脱氢酶和3-磷酸甘油酸激酶的作用。《生物化学杂志》278, 5929–5940.
- Valenti, D., de Bari, L., De Filippis, B., Henrion-Caude, A., and Vacca,R.A. (2014). 线粒体功能障碍作为智力残疾相关疾病的核心角色:唐氏综合征、自闭症、脆性X综合征和Rett综合征概述。《神经科学与生物行为评论》46, 202–217.
- Lin-Hendel, E.G., McManus, M.J., Wallace, D.C., Anderson, S.A., and Golden, J.A. (2016). 放射状与非放射状迁移皮层神经元的差异化线粒体需求:对线粒体疾病的启示。《细胞报告》15, 229–237.
- Rossignol, D.A., and Frye, R.E. (2012). 自闭症谱系障碍中的线粒体功能障碍:系统综述与荟萃分析。《分子精神病学》17, 290–314.
- Slattery, C.F., Beck, J.A., Harper, L., Adamson, G., Abdi,Z., Uphill, J., Campbell, T., Druyeh, R., Mahoney, C.J., Rohrer, J.D., et al. (2014). R47H TREM2变异增加典型早发性阿尔茨海默病风险但不增加朊病毒病或额颞叶痴呆风险。《阿尔茨海默病与痴呆》10, 602–608.e4.
- Csere´p, C., Schwarcz, A.D., Po´sfai, B., La´szlo´, Z.I., Kellermayer, A., Ko¨rnyei, Z., Kisfali, M., Nyerges, M., Lele, Z., Katona, I., et al. (2022). 小胶质细胞通过体嘌呤能连接调控神经元发育。《细胞报告》40, 111369.
- Csere´p, C., Po´sfai, B., and De´nes, A´. (2021). 塑造神经元命运:直接小胶质细胞-神经元相互作用的功能异质性。《神经元》109, 222–240.
- Boumezbeur, F., Mason, G.F., de Graaf, R.A., Behar, K.L., Cline, G.W., Shulman, G.I., Rothman, D.L., and Petersen, K.F. (2010). 通过体内磁共振波谱评估健康衰老中脑线粒体代谢的改变。《脑血流与代谢杂志》30, 211–221.
- Hickman, S.E., Kingery, N.D., Ohsumi, T.K., Borowsky, M.L., Wang, L.C., Means, T.K.,and El Khoury, J. (2013). 直接RNA测序揭示的小胶质细胞感应组。《自然神经科学》16, 1896–1905.
- Go¨tzl, J.K., Brendel, M., Werner, G., Parhizkar, S., Sebastian Monasor, L., Kleinberger, G., Colombo, A.V., Deussing, M., Wagner, M., Winkelmann, J., et al. (2019). PGRN或TREM2缺失导致的相反小胶质细胞激活状态会降低脑葡萄糖代谢。《EMBO分子医学》11, e9711.
- Hyman, B.T., Van Hoesen, G.W., Damasio, A.R., and Barnes, C.L. (1984). 阿尔茨海默病:细胞特异性病理分离海马结构。《科学》225, 1168–1170.
- Busche, M.A., Chen, X., Henning,H.A., Reichwald, J., Staufenbiel, M., Sakmann, B., and Konnerth, A. (2012). 可溶性β淀粉样蛋白在阿尔茨海默病模型小鼠早期海马过度活跃中的关键作用。《美国国家科学院院刊》109, 8740–8745.
- -Si-skova´, Z., Justus, D., Kaneko, H., Friedrichs, D., Henneberg, N., Beutel, T., Pitsch, J., Schoch, S., Becker, A., von der Kammer, H., et al. (2014). 在阿尔茨海默病模型小鼠中树突结构退化与细胞过度兴奋存在功能关联。《神经元》84, 1023–1033.
- Kerchner, G.A., Hess, C.P., Hammond-Rosenbluth, K.E., Xu, D., Rabinovici, G.D., Kelley, D.A.C., Vigneron, D.B., Nelson, S.J., and Miller, B.L. (2010). 采用7-TMRI可视化轻度阿尔茨海默病海马CA1顶端神经毡萎缩。《神经病学》75, 1381–1387.
- Montero-Crespo, M., Dom ´nguez-A´ lvaro, M., Alonso-Nanclares, L., Defelipe, J., and Blazquez-Llorca, L. (2021). 阿尔茨海默病海马CA1区突触组织的三维分析。《脑》144, 553–573.
- Antonucci, F., Turola, E., Riganti, L., Caleo, M.,Gabrielli, M., Perrotta, C., Novellino, L., Clementi, E., Giussani, P., Viani, P., 等 (2012). 小胶质细胞释放的微囊泡通过增强鞘脂代谢促进突触活动。《EMBO杂志》31, 1231–1240.
- Street, K., Risso, D., Fletcher, R.B., Das, D., Ngai, J., Yosef,N., Purdom, E., and Dudoit, S. (2018). Slingshot:单细胞转录组学的细胞谱系和伪时间推断。《BMC基因组学》19, 477.
- Correale, C., Genua, M., Vetrano, S.,Mazzini, E., Martinoli, C., Spinelli, A., Arena, V., Peyrin-Biroulet, L., Caprioli, F., Passini, N., 等 (2013). 髓系细胞表达触发受体细菌传感器调控黏膜炎症反应。《胃肠病学》144, 346–356.e3.
- Mendonc¸ a, P.R.F., Tagliatti, E., Langley, H., Kotzadimitriou, D., Zamora-Chimal, C.G., Timofeeva, Y., and Volynski, K.E. (2022). 低释放效能突触中异步谷氨酸释放增强并分布于整个活性区。《自然通讯》13, 3497.
- Tagliatti, E., Bello, O.D., Mendonc¸ a, P.R.F., Kotzadimitriou, D., Nicholson, E., Coleman, J., Timofeeva, Y., Rothman, J.E., Krishnakumar, S.S., and Volynski,K.E. (2020). 突触结合蛋白1寡聚物钳制并调节神经递质释放的不同模式。《美国国家科学院院刊》117, 3819–3827.
- Bennett, M.L., Bennett, F.C., Liddelow, S.A., Ajami, B., Zamanian, J.L., Fernhoff, N.B., Mulinyawe, S.B., Bohlen, C.J., Adil,A., Tucker, A., 等 (2016). 研究小鼠和人类中枢神经系统小胶质细胞的新工具。《美国国家科学院院刊》113, E1738–E1746.
- Qi, G., Mi, Y., and Yin, F. (2021). 利用急性小鼠脑片打孔技术离体表征脑代谢功能。《Star Protocols》2, 100559.
- 王亮、Chaudhari K、Winters A、太阳、刘锐、杨世豪(2022)。利用Seahorse XFe96分析仪表征啮齿类动物大脑区域特异性葡萄糖代谢谱。《脑血流与代谢杂志》42卷,1259–1271页。
- 郑、G.X.Y.、Terry, J.M.、Belgrader, P.、Ryvkin, P.、Bent, Z.W.、Wilson,R.、Ziraldo, S.B.、Wheeler, T.D.、McDermott, G.P.、Zhu, J.等人(2017)。单细胞的大规模并行数字转录谱分析。Nat. Commun. 8, 14049。
- Hao, Y., Hao, S., Andersen-Nissen, E., Mauck, W.M., Zheng, S., Butler, A., Lee, M.J., Wilk, A.J., Darby, C., Zager, M., 等 (2021). 多模态单细胞数据的整合分析.细胞 184, 3573–3587.e29.
- Cahoy, J.D., Emery, B., Kaushal, A., Foo, L.C., Zamanian, J.L., Christopherson,K.S., Xing, Y., Lubischer, J.L., Krieg, P.A., Krupenko, S.A., 等人 (2008). 星形胶质细胞、神经元与少突胶质细胞的转录组数据库:理解大脑发育与功能的新资源。神经科学杂志 28, 264–278。
- Cembrowski, M.S., Wang, L., Sugino, K., Shields, B.C., 与 Spruston, N. (2016). Hipposeq:海马主要神经元基因表达的全面RNA-seq数据库。eLife 5, e14997。
- 莱恩 E.S.、霍里利茨 M.J.、奥 N.、艾尔斯 M.、本辛格 A.、伯纳德 A.、博埃 A.F.、博古斯基 M.S.、布罗克韦 K.S.、伯恩斯 E.J. 等 (2007)。成年小鼠大脑基因表达的全基因组图谱。自然 445, 168–176。
- Zimmerman, K.D., Espeland, M.A. 与 Langefeld, C.D. (2021). 单细胞研究中伪重复偏倚的实用解决方案。 Nat. Commun. 12, 738.
- Bankhead, P., Loughrey, M.B., Ferna´ndez, J.A., Dombrowski, Y., McArt, D.G.,Dunne, P.D., McQuaid, S., Gray, R.T., Murray, L.J., Coleman, H.G., 等 (2017). QuPath:用于数字病理图像分析的开源软件。科学报告 7, 16878.
- Schafer, D.P., Lehrman, E.K., Heller, C.T., 与 Stevens, B. (2014). 吞噬实验:评估中枢神经系统吞噬细胞与神经元相互作用的实验方案。可视化实验杂志 51482.
¶ 星+方法
关键资源表
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Antibodies | ||
| Monoclonal mouse anti-Tom20 | Santa-Cruz | Cat#sc-17764 |
| Polyclonal rabbit anti-P2YR12 | Anaspec | Cat#AS-55043A |
| Polyclonal guinea pig anti-IBA1 | Synaptic Systems | Cat#234 308 |
| Polyclonal rabbit anti-IBA1 | WAKO Chemicals | Cat#019-19741 |
| Polyclonal guinea pig anti-DCX | Merck-Milipore | Cat#AB2253 |
| Polyclonal guinea pig anti-MAP2 | Synaptic Systems | Cat#188 004 |
| Polyclonal rabbit anti-vGlut1 | Synaptic Systems | Cat#135 302 |
| Monoclonal rat anti-CD68 | Biolegend | Cat#137001 |
| Polyclonal rabbit anti-Neurod2 | Abcam | Cat#ab104430 |
| Polyclonal rat anti-Ctip2 | Abcam | Cat#ab18465 |
| Monoclonal rat anti-CD45 Pe | Biolegend | Cat#103106 |
| Monoclonal rat anti-CD45 PerCP | Biolegend | Cat#103130 |
| Monoclonal rat anti-CD11b Pe-Cy7 | Biolegend | Cat#101216 |
| Monoclonal rat anti-CD16.32 | Biolegend | Cat#101301 |
| Monoclonal mouse anti-NeuN 488 | Merck-Milipore | Cat#MAB377X |
| Anti-Puromycin 647 | Scenith -Auguello Lab | RRID:AB_2827926 |
| Polyclonal rabbit anti-PSD95 | Thermo-Fisher | Cat#51-6900 |
| Polyclonal rabbit anti-Trem2 Goat anti-Mouse IgG (H+L) Highly | R&D Systems | Cat#AF1729 |
| Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | Thermo-Fisher | Cat#A11031 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo-Fisher | Cat#A11034 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody Alexa Fluor 633 | Thermo-Fisher | Cat#A21071 |
| Goat anti-Rat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Thermo-Fisher | Cat#A21247 |
| Goat anti-Guinea Pig IgG(H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 Goat anti-Guinea Pig IgG (H+L) Highly | Thermo-Fisher | Cat#A11073 |
| Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 Goat anti-Guinea Pig IgG (H+L) Highly | Thermo-Fisher | Cat.#A11075 |
| Cross-Adsorbed Secondary Antibody, Alexa Fluor 633 Donkey anti-Sheep IgG (H+L) Highly | Thermo-Fisher | Cat#A21105 |
| Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo-Fisher | Cat#A-11015 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | Thermo Fisher | Cat#A-10042 |
| Fluorsave | Millipore | Cat. #345789 |
| Continued REAGENT or RESOURCE | ||
| Chemicals,peptidesandrecombinantproteins | SOURCE | IDENTIFIER |
| CNQX disodium salt | Tocris | Cat#1045/1 |
| D-AP5 | Tocris | Cat#0106/1 |
| FCCP | Agilent Technologies | Cat#103015-100 |
| Oligomycin | Agilent Technologies | Cat#103015-100 |
| Rotenone/Antimycin A | Agilent Technologies | Cat#103015-100 |
| Puromycin | Scenith/Auguello Lab | N/A |
| 2-Deoxy Glucose | Scenith/Auguello Lab | N/A |
| Oligomycin | Scenith/Auguello Lab | N/A |
| Neuromag reagent Epoxy resin Poly/Bed@ 812 | OZ-Biosciences Polysciences | Cat#KC30800 Cat#08792-1 |
| 1% Uranyl Acetate | Electron Microscopy | Cat#22400-1 |
| 4% Osmium Tetroxide | Sciences (EMS) Electron Microscopy | Cat#19140 |
| Propylene Oxide | Sciences (EMS) | |
| Hoechst-33342 | TAAB Thermo-Fisher | Cat#P021 |
| BioTracker ATP-Red Live Cell Dye | Merck-Millipore | Cat#62249 |
| MitoTracker Deep Red FM | Thermo-Fisher | Cat#SCT045 Cat#M22426 |
| CellRosGreen reagent | Thermo Fisher | Cat#C1044 |
| Anti-Mouse Ig,kK/Negative Control CompensationParticles Set | BD Biosciences | Cat#552843; RRID: |
| Anti-RatIg,/egativeCotrol | BD Biosciences | AB_10051478 Cat#552844; |
| Compensation Particles Set Critical commercial assays | RRID: AB_10055784 | |
| Seahorse XF Cell Mito Stress Test Kit Agilent Technologies | ||
| Fixation/Permeabilization Solution Kit | BD Bioscience | Cat#103015-100 |
| Cat#554714 | ||
| Foxp3 /Transcription Factor Staining Bufer Set | Thermo-Fisher | Cat#00-5523-00 |
| Zombie NIRrm Fixable Viability Kit | Biolegend | Cat#423105 |
| Chromium Next GEM Single Cell3' Kit v3.1 | 10X Genomics | Cat# PN-1000269 |
| Chromium Next GEM Chip G Single Cell Kit | 10X Genomics | Cat# PN-1000127 |
| Dual Index Kit TT Set A | 10X Genomics | Cat# PN-1000215 |
| Direct-ZolTM MiniPrep Isolation Kit | Zymo Research | Cat# R2050 |
| High-Capacity cDNA RT kit | Applied Biosystems | Cat# 4368814 |
| TaqMan Fast Universal PCR Master | Applied Biosystems | Cat# 4352042 |
| Mix(2x), no AmpErase UNG | ||
| Deposited data | ||
| scRNA-seq data Experimental models: Organisms/strains | This Paper | Geo number: GSE249036 |
| Mouse C56BL/6J | ||
| mouse: Trem2-/- | Charles River | Strain Code 632 |
| Recombinant DNA | Turnbull et al.15 | N/A |
| pFU_GFP | Kirill Volynski, UCL | N/A |
| pAAV.hSynap.SF-iGluSnFR.A184V | Kill Volynski, UCL | Cat#106175; RRID: Addgene_106175 |
| Software and algorithms | ||
| GraphPad Prism 9 | GraphPad | https://ww.graphpad.com; RID:SCR_002798 |
| Fj Adobe Ilustratr CC | NIH Adobe | https://fjisc; RRID:SCR_002285 http://www.adobe.com/products/ilustrator.html |
| RRID:SCR_010279 | ||
| Radius 2.0 software | EMSIS | https://www.emsis.eu/products/radius 百继续 |
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Seahorse Wave Version | Agilent Technologies | http://www.agilent.com/en-us/products/ cell-analysis-(seahorse)/software- download-for-wave-desktop; RRID:SCR_014526 |
| MetaFluor Fluorescence Ratio Imaging Software | Molecular Devices | RRID:SCR_014294 |
| Matlab R2018 or R2021b | Mathworks | https://t.mathworks.com/; RRID: SCR_001622 |
| FlowJo 10 | FlowJo | https://www.flowjo.com/; RRID: SCR_008520 |
| Bcl-convert 3.8.4 | Bcl-convert | https://emea.support.illumina.com/ sequencing/sequencing_software/ bcl-convert.html |
| 10x Genomics Cell Ranger 6.1.1 | Cellranger | https://www.10xgenomics.com/; RRID:SCR_023221 |
| R 4.2.2 | R | https://www.r-project.org/; RRID: SCR_002394 |
| Seurat 4.2.0 | Seurat | https://satijalab.org/seurat/articles/ get_started.html; RRID:SCR_016341 |
| Bioconductor Milor 1.2.0 | MiloR | https://www.bioconductor.org/packages/ release/bioc/html/miloR.html |
| Slingshot 2.6.0 | Slingshot | https://www.bioconductor.org/packages/ release/bioc/html/slingshot.html106 |
| pClamp-10 | Axon Instruments, | https://www.moleculardevices.com; RRID:SCR_011323 |
| BrainWave 4 | Foster City, CA 3Brain AG, Switzerland | https://www.3brain.com/products/ |
| Biorender | Biorender | software/brainwave4 http://biorender.com: RRID:SCR_018361 |
| Via7 software system | Applied Biosystems | http:https://www.thermofisher.com/it/ en/home/life-science/pcr/real-time-pcr/ real-time-pcr-instruments/viia-7-real- |
| Imaris v. 9.7.2 or 7.2.3 | Oxford Instruments | time-pcr-system.html RRID:SCR_023358 http://www.bitplane.com/imaris/imaris RRID:SCR_007370 |
| QuPath v0.3.2 | QuPath | https://doi.org/10.1038/ s41598-017-17204-5 |
¶ 资源可用性
主要联系人更多信息及资源和试剂的请求应直接发送给主要联系人 Michela Matteoli (michela.matteoli@hunimed.eu),并将由其负责处理。
¶ 材料可用性
本研究未产生新的独特试剂。
¶ 数据和代码可用性
单细胞RNA-seq数据已存入GEO数据库,并于出版当日公开。登录号列于关键资源表中。d 本文未报告原始代码。d重新分析本文报告数据所需的任何补充信息,均可根据要求向主要联系人获取。
¶ 方法细节
¶ 老鼠
所有实验均根据欧洲共同体理事会(2010年9月22日第2010/63/EU号指令)和意大利第26/2014号立法法令制定的指南进行。本研究经Humanitas研究医院的机构动物护理与使用委员会(IACUC)及意大利卫生部批准。C57BL/6J 小鼠(制备方法如先前报道15)由Bioxell-Cosmo Pharmaceutical(意大利米兰)提供107。C57BL/6J小鼠购自Charles River实验室。小鼠饲养于无特定病原体环境中,恒定温度( )和湿度( ),12小时光照/黑暗循环,自由获取食物和水。除特别说明外,各实验均使用出生0-1天(P0-1)及18-20天(P18-20)的雄性与雌性小鼠。
¶ 原代海马神经元培养与转染
从或的P0-1小鼠15中分离原代海马神经元,并在补充了 B27(货号17504044,赛默飞世尔科技)的完全NeurobasalA培养基(货号10888,赛默飞)中培养。简言之,将海马组织解剖后采用 胰蛋白酶在 下酶消化15分钟进行解离,随后使用标准p1000微量移液器进行机械吹打。将神经元以每盖玻片10万至12万细胞的密度铺设在多聚赖氨酸( ;货号P2636,西格玛奥德里奇)包被的18毫米或24毫米直径玻璃盖玻片上(用于成像和电生理实验),或以每孔12000细胞的密度接种于经多聚赖氨酸( )处理的XF-96细胞培养板卡匣中(用于海马细胞外通量分析)。实验在体外培养4-5天或14-16天时进行。在活体成像和免疫荧光染色中,海马神经元于培养第5天使用Neuromag试剂(货号KC30800;OZ Biosciences)分别转染pFU_GFP或pAAV.hSynap.SF-iGluSnFR.A184V质粒。该方法使得iGluSnFR探针仅在少量神经元亚群( 至 )中表达,这对单个突触终末的囊泡释放成像至关重要。pFU_GFP与pAAV.hSynap.SF-iGluSnFR.A184V质粒由英国伦敦大学学院的K. Volynski教授惠赠。
¶ 原代小胶质细胞培养
原代小胶质细胞培养取自混合胶质细胞培养物,方法如前所述43,44。简要流程如下:剥离P2新生小鼠脑膜后,在冷的汉克斯平衡盐溶液(HBSS,货号14180046,赛默飞世尔)中轻柔分离大脑皮层与海马体。经 胰蛋白酶消化及机械分离后,将细胞接种于T-75培养瓶,并在含有 热灭活胎牛血清(FBS,货号ECS0186L,Euroclone)、 葡萄糖、 丙酮酸钠(货号ECM9542D,Euroclone)及 青霉素/链霉素(货号15140-122,赛默飞世尔)的EMEM培养基(货号M5650,Sigma-Aldrich)中培养。约在第15天体外培养时,通过245转/分钟振荡培养瓶60分钟分离获得小胶质细胞。
在条件培养基制备过程中,小胶质细胞以每孔 个细胞的密度,接种于经多聚鸟氨酸(货号P8638,Sigma Aldrich)预处理的6孔板中,培养基于Neurobasal-A/B27培养基。体外培养24小时后,收集条件培养基(CM),以 离心20分钟,随后用于后续实验。
¶ 活体成像实验
¶ 4天体外培养神经元中的ATP成像
从和新生幼鼠获取的4DIV原代海马神经元,在 的Neurobasal-A培养基中加载线粒体ATP生物追踪剂ATP-red活细胞染料(货号SCT045,Merk-Millipore)45分钟,随后进行成像以测量线粒体ATP产量。通过稀疏GFP转染或神经元胞体的明场图像识别体细胞神经元。神经元持续置于细胞外缓冲液1(EB1)中,该缓冲液包含 NaCl、 、 、2 mM CaCl2、 葡萄糖和 Hepes( )。108,109在记录基础ATP-red荧光2分钟后,向细胞施加线粒体复合物1抑制剂鱼藤酮与抗霉素A混合液( ,货号103015-100,安捷伦科技),并继续监测ATP衰减10分钟。ATP动态变化的时间推移记录以 采集速率进行。将18毫米记录腔室(货号QR-40LP,Warner Instruments)置于配备EMCCD相机(QuantEM-SC 512x512,Photometrics)的IX-71倒置显微镜(奥林巴斯)载物台,采用发光二极管光源(Cairn research,Optoled Lite)并通过采集软件(MetaFluor,MolecularDevices)控制照明以减轻光漂白。使用40倍油镜(数值孔径1.3)识别视场内多个神经元胞体( 像素)。ATP衰减量以ΔF/F0表示,通过测量ΔFrotenone确定线粒体ATP产量,所有数值均以神经元为基准进行标准化。
¶ 14DIV神经元培养中的双色突触成像
从和新生幼鼠获取的14-16天海马神经元经iGluSNFR探针转染后,在Neurobasal-A培养基中与线粒体ATP生物追踪剂ATP-red活细胞于 共同孵育45分钟。首先以10赫兹频率给予5次刺激激活神经元,通过绿色通道记录单个突触终末的谷氨酸释放量。经过5分钟恢复期后,在同一突触终末的红色通道记录突触ATP-red信号。为诱发突触线粒体ATP生成,以100赫兹频率施加200次刺激挑战神经元,并记录10分钟内的ATP动态变化。双色成像实验在 2 3 . 2 5 ^ { \circ } ^ { \circ } \mathrm { C } 的开放式层流场刺激腔(型号QR/RC-47FSLP,沃纳仪器公司,哈姆登,CT)中进行。细胞外缓冲液(EB1)成分为 、 KCl、 、2 mM CaCl2、 3 0 \mathrm
葡萄糖和 Hepes( )108,109。为阻断重复放电活动,在EB1中添加6-氰基-7-硝基喹喔啉-2,3-二酮(CNQX)( ;目录号479347-85-8,Tocris)和DL-AP5( ;目录号79055-68-8,Tocris)。通过连接刺激隔离单元(SIU-102;Warner Instruments,哈姆登,CT)的序列发生单元(Digitimer Ltd, DG2A)施加振幅 、时长1毫秒的脉冲来诱发电刺激响应。记录腔放置于配备EMCCD(电子倍增CCD)相机(QuantEM-SC 512x512,Photometrics)的IX-71倒置显微镜(Olympus,汉堡,德国)载物台上。采用发光二极管LED(Cairn research, Optoled Lite)作为照明光源,并与采集软件(MetaFluor, Molecular Devices)联用以减少光漂白。使用40倍(数值孔径1.3)油浸物镜识别iGluSNFR阳性神经元的轴突分支。在 毫米( 像素)的感兴趣区域(ROI)内记录诱发的iGluSNFR或ATP-red响应,该区域通常包含20至100个单个突触。谷氨酸动态变化的时序记录采集速率为 ,持续 秒;而线粒体ATP记录采集速率为 ,持续12分钟。使用ImageJ(NIH)和MATLAB(MathWorks)定制软件脚本分析图像109。通过从5个动作电位10赫兹爆发后的峰值荧光中减去静息iGluSNFR荧光来识别谷氨酸突触响应,随后生成 毫米ROI用于测量每次实验中所有已识别突触的平均荧光响应。扣除背景后,将数据标准化为静息iGluSNFR或ATP-red信号(ΔF/F0)。
¶ 海马细胞外通量分析在原代培养物中的应用
从和新生幼鼠获取的原代海马神经元及培养小胶质细胞的生物能量学特性,通过XF-96海马细胞外通量分析仪(安捷伦科技有限公司,加州)进行测定。神经元与小胶质细胞在 、 条件下分别培养至4天(神经元)或24小时(小胶质细胞)。在指定时间点,将神经元和小胶质细胞清洗后置于XF检测培养基(XF DMEM pH 7.4,补充 葡萄糖、 丙酮酸钠和2 M L-谷氨酰胺,货号103575/103578/103577/103579,安捷伦科技)中,根据制造商方案进行线粒体压力测试呼吸测量。实时获取以下条件下的测量数据:无药物处理(基础条件)及顺序注射 寡霉素、 FCCP、 鱼藤酮加抗霉素A(Rot/AA,货号103015-100,安捷伦科技)。耗氧率数据以培养基中 消耗的皮摩尔值表示,并相对于对照组进行标准化。未加药基础条件下的OCR测量值代表基础OCR;FCCP注射后的OCR测量值代表最大OCR;寡霉素注射后的OCR测量值代表ATP偶联呼吸;Rot/AA注射后的OCR测量值代表非线粒体残余呼吸。每个条件进行三次测量循环,每次实验每个条件至少设置15个重复。使用GraphPad Prism软件对基础条件及加药后的OCR值进行作图并开展统计分析。
¶ 离体荧光激活细胞分选测量
P1海马体在立体定位显微镜下于冰冷的HBSS中被仔细解剖,并在冰上使用冷缓冲液在 下进行机械消化,如先前所述.110 胞首先与FC阻断剂CD16.32(货号101301,克隆93,1:100,Biolegend)孵育15分钟,然后在FACS缓冲液( FBS,1 mM EDTA于磷酸盐缓冲盐水中)中与以下荧光标记抗体孵育30分钟于冰上:CD45-Pe或PerCP(货号103106和103130,克隆30-F11;1:500,Biolegend)、CD11b-Pe-Cy7(货号101216,克隆M1-70;1:200,Biolegend)。表面染色后,样品用FACS缓冲液洗涤两次,并在 下以300xg离心5分钟。随后进行活性染料染色15分钟,通过将样品重悬于含有Zombie Nir(货号423105,1:800,Biolegend)的磷酸盐缓冲盐水(PBS,货号TL1006,Microgem)中。对于细胞内染色,使用了FOXP3缓冲试剂盒(货号00-5523-00,Thermo-Fisher)。透化和固定,以及随后的洗涤步骤、孵育和离心,均按照方案进行。细胞内染色在冰上进行1小时,使用了以下抗体:抗-NeuN 488(货号MAB377X,克隆A60;1:300,Merck-Millipore)、抗-嘌呤霉素 AF647(货号AB_2827926,克隆R4743L-E8;1:400,Auguello Lab)。对于代谢染色,细胞与CellROXgreen(货号C1044,Thermo-Fisher)或Mitotracker Deep Red FM(货号M22426,Thermo-Fisher)在 下孵育35分钟。代谢染色未与细胞内染色“耦合”。FACS分析在FACS Fortessa机器或FACS Canto II(BD Biosciences)上进行,数据使用FlowJo软件(TreeStar)进行分析。
¶ 通过翻译抑制测定分析单细胞能量代谢
SCENITH检测依据Arg€uello等人49的方法进行,并稍作修改(详见www.scenith.com)。海马体分离后采用木瓜蛋白酶消化法(cat.LK003153,Worthington Biomedical)在 持续振荡条件下消化45分钟。用含 FBS的Neurobasal-A培养基终止反应后,将消化后的海马组织用P1000移液器轻柔匀浆,经 滤网过滤,室温 离心10分钟。所得细胞悬液重悬后以150,000细胞/孔的密度接种于96孔板。细胞在 、 条件下恢复45分钟,随后与不同代谢抑制剂混合物在 、 环境中孵育10分钟。使用的代谢抑制剂包括: -脱氧葡萄糖、 寡霉素、2-脱氧葡萄糖与寡霉素联用(浓度同上,Scenith)。接着向样本中加入 嘌呤霉素,在 、 条件下继续孵育45分钟。最后用FACS缓冲液清洗细胞两次,于 以 离心5分钟,并按照前文所述进行流式细胞术分析。
¶ 海马切片中海马鱼细胞外物质转运分析
从和的P1或P18日龄雄性后代获取的离体急性脑切片,其生物能量学特性采用XF-96海马细胞外通量分析仪(Seahorse Bioscience,CA)并通过调整后的实验方案进行测定。111,112 小鼠被通过 吸入深度麻醉后断头。迅速取出大脑并立即浸入预先用 和 饱和至少15分钟的冰切解剖aCSF溶液中。该aCSF溶液包含(单位mM):129氯化钠、1.25磷酸二氢钠、10葡萄糖、1.8硫酸镁、1.6氯化钙、3氯化钾、21碳酸氢钠。使用VT1000S振动切片机(Leica Biosystems Nussloch GmbH)切割300毫米冠状海马切片,在 的高蔗糖保护溶液中于 环境下保存至少60分钟,以备后续处理使用。
在组织次级处理中,使用1毫米快速取样打孔器(Ted Pella公司)手动获取覆盖P1阶段海马CA区、P18脑切片CA1与CA3亚区的1毫米直径300微米厚组织样本。样本迅速浸入含氧测定培养基(单位mM: 、5 KCl、 、 、 、25 HEPES、0.01丙酮酸盐和6葡萄糖, \mathsf { p H } 7 . 4 \AA , )后,以单样本/孔形式接种至预先包被多聚赖氨酸( )的XF-96细胞培养板。样本经清洗后使用XF测定培养基进行线粒体压力测试呼吸测量。实时测量分为无药物处理(基础条件)与序贯注射 寡霉素、 FCCP、 鱼藤酮加抗霉素A(Rot/AA,货号103015-100,安捷伦科技)两个阶段。耗氧率数据以培养基中 消耗的皮摩尔数表示,并在各实验内对照条件下标准化。鉴于切片组织动力学较慢,多数条件采用8个测量循环。无药物基础条件下的OCR代表基础耗氧率;FCCP注射后OCR代表最大耗氧率;寡霉素注射后OCR代表ATP耦合呼吸;Rot/AA注射后OCR代表非线粒体残余 消耗。将基础条件及药物添加后的OCR值在各实验内相对于对照组标准化,使用GraphPad Prism进行绘图与统计分析。基础呼吸值低于建议范围( 分钟)或对Seahorse药物无响应的样本均予以剔除。
¶ RNAseq分析
¶ 样品制备
从3个独立窝次的和幼鼠中取P1海马体(每窝每次样本 个胚胎),在冰冷HBSS溶液中快速解剖后混合汇集。实验采用对照组和小鼠中雌雄数量均衡的混合样本,以最大限度减少性别因素影响。
使用木瓜蛋白酶 dissociation 系统试剂盒(Worthington,货号#LK003150)对解剖组织进行解离,操作遵循制造商说明书及前期优化方案。解离后的海马组织悬浮于含 BSA(Sigma Aldrich)的PBS缓冲液中。每个样本约8,000个细胞通过Single Cell 单细胞试剂盒(10X Genomics)加样至单细胞芯片G的单个通道中,在Chromium系统内生成凝胶珠乳液。完成细胞捕获与裂解后,按照制造商(10X Genomics)protocol进行cDNA合成并扩增14个循环。取50 ng扩增后cDNA用于各样本的Illumina测序文库构建。测序在NextSeq2000 Illumina测序平台完成,遵循 Genomics的读长生成指引。
¶ 单细胞测序分析
原始测序数据使用Illumina bcl-convert工具转换为fastq文件,该工具集成在CellRanger(10X Genomics)套件(版本6.1.1.113)中。从原始数据开始,使用CellRanger分析流程生成数字基因表达矩阵。使用预构建的小鼠基因组(版本 \mathrm { m m l } 0 { - } 1 . 2 . 0 \AA \AA )作为基因组参考。采用CellRanger count模块进行读数比对,默认设置下将序列长度设为r1长度 和r2长度 。每个样本的每个细胞至少产生35,000条读数。通过Seurat R软件包 将原始数字基因表达矩阵(每个细胞每个基因的UMI计数)导入R 4.4.2版本。简言之,将每个生物学重复的每细胞每基因UMI计数导入Seurat后,通过以下标准进行样本质量控制:过滤表达独特基因数少于1,000个、UMI总数低于1,000个、或线粒体基因比对率超过 的细胞。每个时间点的数据均进行细胞特征表达标准化,将其总表达量乘以10,000的比例因子后,对结果进行 转换。随后根据G2/M期和S期标志物的表达为单个细胞分配细胞周期评分。通过FindVariableFeatures函数(选择方法 “vst”,特征数 )识别前2,000个变异基因,对标准化后的表达值进行缩放。基于缩放数据执行主成分分析(PCA)线性降维,采用基于图的聚类方法进行细胞分群(RunPCA)。对各独立样本进行SCT标准化(负二项回归)时不排除任何变量(SCTransform(seurat, 需回归变量 NULL, 仅返回变异基因 。针对性别导致的细胞聚类差异,通过Seurat内置的SelectIntegrationFeatures(特征数 )、PrepSCTIntegration、FindIntegrationAnchors(标准化方法 “SCT”)及IntegrateData函数进行数据整合。采用基于图的方法(肘部图)确定主成分数量,使用Louvain算法(Seurat内置)进行迭代细胞聚类,通过分析聚类树图(clustree)选择最佳分辨率。通过Seurat内置的FindAllMarkers函数(经Bonferroni校正的Wilcoxon秩和检验;校正后P值 )鉴定差异表达基因进行聚类注释,仅检测在簇内至少 细胞中表达且与剩余细胞存在平均0.25倍(对数尺度)差异的基因。通过综合分析所得标志基因、经典标志基因及文献中的附加标志基因115–117,最终确定细胞类型身份。随后滤除血管细胞、间充质细胞和室管膜细胞,对剩余细胞按前述方法重新进行聚类分析。为了测试基因型之间的差异细胞丰度,我们在潜在空间嵌入生成的KNN图上使用MiloR 1.2.0功能,采用默认参数,并将“基因型”作为协变量。
¶ 细胞类型特异性差异基因表达分析
为了确定差异表达基因,我们使用了Seurat功能中的FindMarkers,采用默认的两部分障碍模型(MAST)实现,添加了随机效应变量(MAST-re测试; 调整后的 ,仅测试在簇内至少 细胞中发现的基因,并且平均显示出在基因型间至少有0.10倍的差异(对数尺度),并将批次设置为随机效应变量。CA-ImmPyr、CA1-Pyr和CA3-Pyr细胞类型的差异表达基因列表使用Ingenuity Pathway Analysis(IPA - Qiagen Ingenuity Systems) 的经典通路富集进行了分析。
¶ 轨迹分析
伪时间和轨迹树是通过Slingshot软件包分两步推断得出的。首先,我们基于聚类的最小生成树(MST)识别全局谱系结构;随后利用slingshot函数在UMAP空间中拟合同步主曲线以描述各谱系。轨迹推断与可视化在整合预处理数据对象的UMAP空间中进行,并采用预先标注的细胞类型标签。将顶端祖细胞(AP)设为轨迹的起始根节点,所有推断出的伪时间变量(每个谱系独立计算)均被分别添加至元数据中。接下来,我们通过从每个谱系随机抽取100个细胞,并对经验累积分布函数进行双侧Kolmogorov-Smirnov检验,比较三个谱系间细胞的伪时间分布。最后,我们重复相同流程,针对每个谱系中的不同细胞类型,按基因型分别比较细胞的伪时间分布。
¶ RNA提取和qRT-PCR
从P1、P20、P90冷冻海马体或 P18小鼠CA1/CA3区域采集的1毫米组织样本,在冰上温和解冻后,使用自动组织研磨仪配合匀浆珠,置于500毫升TRI Reagen ^ \mathrm { { \textregistered } } (Zymo Research)中进行机械破碎。随后按照制造商指南,采用RNA Direct-Zol MiniPrep分离试剂盒(Zymo Research)进行RNA提取。最终将RNA溶于25毫升无DNA酶/RNA酶水中,并使用NANOdrop 2000c分光光度计(Thermo Fisher Scientific)检测RNA浓度及 吸光度比值。
每个条件下的 RNA使用高容量cDNA反转录试剂盒(Applied Biosystems)进行反转录合成cDNA。采用TaqMan检测试剂盒(TaqMan Fast Universal PCR Master Mix ,不含AmpErase UNG,ThermoFisher)在qRT-PCR Viia7软件系统(Applied Biosystems)上进行实时定量聚合酶链式反应(qRT-PCR),反应终体积为 。每个基因至少进行重复检测,数据分析采用比较DCT法。各目标基因的mRNA检测值均以内参基因Gapdh进行标准化。使用的TaqMan检测探针如下(Applied Biosystems):小鼠Trem2 FAM-MGB Mm04209424_g1;小鼠GAPD(GAPDH) VIC-MGB内参对照。
¶ 电生理学
¶ 原代神经元培养
对来自和 基因型P1小鼠的培养4天原代海马神经元进行全细胞电压钳记录。实验过程中,神经元始终置于含细胞外缓冲液2的18毫米记录槽(型号QR-40LP,华纳仪器公司,康涅狄格州哈姆登)中。细胞外缓冲液(EB2)成分为(单位mM):125NaCl、5 KCl、 、 、 、6葡萄糖及25 HEPES-NaOH( )。记录电极(电阻 )内充标准细胞内液,含(单位 ): -葡萄糖酸盐、1 EGTA、10 HEPES、 、4 MgATP及0.3 Tris-GTP( )。电压依赖性电流激活实验中,神经元钳制电位为 ,并施加从- 起始至 的递增去极化阶跃电压( )。钠电流密度通过峰值内向电流除以细胞电容( )计算。静息电位在电流钳模式下于 时测定,输入电阻则在电压钳模式下通过不同去极化阶跃电压(100至- )稳态电流的I/V关系曲线斜率计算。记录使用Multiclamp 700B放大器及pClamp-10软件(Axon Instruments,加州福斯特城)在电压钳模式下完成。串联电阻范围为10-20 MΩ并在整个记录过程中持续监测。信号经放大后以 采样,2-3 KHz滤波,采用pClamp 10数据采集分析系统处理。培养体系中漏电流超过 pA的细胞被排除分析。
¶ 脑切片上的多电极阵列 (MEA)
P18 和雄性小鼠通过 吸入深度麻醉后断头处死。取出脑组织并立即浸入预饱和15分钟以上( O2与 CO2混合气)的冰切aCSF溶液中。该aCSF溶液成分为(单位mM): 、 、10葡萄糖、 、 、3 KCl、 . 使用VT1000S振动切片机(Leica Biosystems Nussloch GmbH)切割400微米厚冠状海马切片,置于 高蔗糖保护液中4 保存至少60分钟后用于记录。记录过程中将切片置于多电极阵列上,以2毫升/分钟流速持续灌注aCSF溶液。切片放电活动稳定15分钟后,继续采集5-10分钟自发胞外信号。
所有细胞外多位点记录均采用CMOS生物传感器及采集系统(瑞士3Brain AG公司)完成。实验使用了高密度多电极CMOS阵列芯片,该芯片采用 阵列配置,集成4096个记录电极。数据采集通过BrainWave 4软件(瑞士3Brain AG公司)进行控制。原始数据以18千赫兹的采样率实现数字化。
每次实验均采集记录单元中切片的图像,仅保留位于CA1和CA3区域的电极。对区域进行了分析。所有MEAs分析算法均采用定制编写的MATLAB脚本。首先,噪声信号我们识别并从分析中排除。然后,使用BrainWave软件(3Brain Gmbh,瑞士)的二阶巴特沃斯滤波器对记录进行高通滤波( )。采用了一种尖峰检测算法(精确时序尖峰检测,PTSD)来从 信号中提取尖峰时间。设置了7个标准差的阈值以滤除基底噪声。
在不应期内(设定为1毫秒)出现的尖峰信号已被剔除。利用每个检测到神经元的时标数据生成点阵图。通过将尖峰总数除以记录时长,计算得出单个神经元的平均放电频率。为评估海马各亚区间的相互作用,采用MATLAB的xcorr函数计算了合适的互相关指标。通过计算零滞后时的最大相关系数并构建相关性矩阵,进而估算每次实验的平均相关系数,以此判定不同神经元放电模式间的同步化程度。而所分析的神经元数量,则对应各海马亚区采样的有效电极数量。
¶ 免疫荧光分析
¶ 海马切片
P1幼鼠通过断头处死,迅速取出脑组织并在新鲜PBS1X缓冲液中冲洗,随后于 多聚甲醛(PFA)中固定过夜。P18大鼠经甲苯噻嗪/氯胺酮混合剂深度麻醉后,行经心灌注 PFA固定,并额外进行过夜后固定。使用VT1000S振动切片机(莱卡显微系统)切割50微米厚冠状脑片。脑片经冷PBS1X冲洗后,分别采用 地高辛-PBS1X溶液通透15分钟或 TritonX-100-PBS1X溶液通透1小时,随后用含 正常山羊血清(NGS) 牛血清白蛋白(BSA)/驴血清(DS)与 TritonX-100的PBS1X封闭液孵育30分钟以阻断非特异性结合位点,最后于 条件下与一抗共同孵育过夜。
随后将切片用PBS清洗3次,并与偶联Alexa Fluor(488、555或633)的二抗共同孵育。使用以下一抗:豚鼠抗IBA1(货号234 308,1:1000,Synaptic Systems)、兔抗IBA1(货号019-19741,1:1000,和光纯药)、兔抗P2yr12(货号AS-55043A,1:400,Anaspec)、小鼠抗Tom20(货号sc-17764,1:200,Santa Cruz)、豚鼠抗DCX(货号AB2253,1:500,Merck-Millipore)、豚鼠抗MAP2(货号188 004,1:1000,Synaptic Systems)、大鼠抗CD68(货号137001,1:1000,Biolegend)、兔抗PSD95(货号51-6900,1:200,Thermo-Fisher)、绵羊抗Trem2(货号AF1729,1:200,R&D Systems)、兔抗Neurod2(货号ab104430,1:1000,Abcam)、大鼠抗Ctip2(货号ab18465,1:100,Abcam)。所有切片均用Hoechst-33342(货号62249,1:1000,Thermo-Fisher)复染,并用Fluorsave(货号345759,Millipore)封片。 使用配备HC PL APO CS2物镜及ACS APO 或 油浸物镜的Leica SP8I或SP8II激光扫描共聚焦显微镜获取图像。Tom20分析中,采用 或 油浸物镜获取整个锥体层(P1)或海马CA1与CA3亚区区域(P18)。通过基于CA1和CA3锥体层Dcx或Map2染色生成二值掩膜来估算Tom20荧光强度,并按面积分数标准化。图5相关染色使用来自图3和图4的对照组动物子集。通过QuPath软件中的阳性细胞检测功能计数CA1和CA3锥体层NeuroD2阳性细胞以估算神经元密度。119 同样,通过计数Iba1阳性细胞并除以CA1和CA3面积来估算小胶质细胞密度。使用Fiji软件(NIH,贝塞斯达)的Skeleton插件评估海马小胶质细胞形态,该插件可对组织中的骨架化小胶质细胞进行三维形态分析。通过基于3D Iba1标记生成二值掩膜来测量单个海马小胶质细胞的P2y12r强度。采用Imaris软件(牛津仪器,v. 9.7.2)进行三维表面重建和体积量化,以评估小胶质细胞吞噬功能与Trem2免疫反应性。 120 PSD95强度以整张图像的体积分数表示。对于PSD95吞噬定量分析,仅考虑与小胶质细胞内CD68共定位的信号。
¶ 原代海马神经元
将4DIV或14DIV神经元在PBS1X溶液和蔗糖溶液中用 多聚甲醛固定20分钟,再用含 皂苷的PBS1X溶液透化处理10分钟。在室温下与一抗孵育2小时前,先用含 正常山羊血清和 皂苷的PBS1X溶液封闭非特异性结合位点30分钟。神经元经PBS1X溶液洗涤3次后,与Alexa Fluor(488、568或633)标记的二抗在室温下孵育45分钟。盖玻片经PBS1X溶液洗涤3次,用Hoechst复染显示细胞核,最后用Fluorsave封片剂封固于载玻片。使用的一抗包括:小鼠抗Tom20(货号.sc-17764;1:200,Santa Cruz);豚鼠抗Map2(货号188004;1:1000,Synaptic Systems)以及兔抗VGlut1(货号135302;1:1000,Synaptic Systems)。采用ImageJ(NIH)定量分析胞体与神经突或突触的荧光强度:通过手动追踪胞体和初级神经突来评估经背景信号校正的Tom20胞体与神经突荧光强度,而Tom20突触强度则通过基于vGlut1染色叠加GFP阳性神经元生成的二值掩膜进行量化。
对4DIV稀疏分布神经元进行Sholl分析时,我们采用Fiji平台的Sholl分析插件。简而言之,以转染细胞体中心为原点绘制一系列半径递增的同心圆,通过统计树突分支与各圆周的交点数量来评估神经元树突分支的复杂程度。图像采集使用配备ACS APO 40倍或63倍油镜的徕卡SP8I共聚焦显微镜完成。
¶ 电子显微镜 P18 切片
和雄性小鼠在P18日龄经甲苯噻嗪/氯胺酮混合剂深度麻醉后,进行经心脏灌注新鲜PBS1X溶液。取出脑组织并置于冰镇溶液中,该溶液包含以下成分(单位mM): 、 、10葡萄糖。
, ,3 KCl, (, ,用 和 平衡。使用VT1000S振动切片机(Leica Microsystems)从内侧前额叶皮层(PFC)切取 厚冠状切片。切片在冷PBS1X中冲洗后,于室温下用含 戊二醛的 二甲胂酸盐缓冲液固定3小时。随后在室温下进行后固定:先于 锇酸(货号19140,Electron Microscopy Science,Hatfield, PA, USA)中固定2小时,再于 醋酸铀水溶液(货号22400-1,Electron Microscopy Science)中固定1小时。
随后将切片通过梯度乙醇系列脱水,以环氧丙烷作为过渡液(产品编号P021,TAAB Laboratories Equipment,英国奥尔德马斯顿),并在 下包埋于环氧树脂(Poly-Bed;产品编号08792-1,Polysciences,美国沃灵顿)中过夜,最终在 下固化2天。对目标区域(海马CA1和CA3区)的后续观察在200纳米半薄切片上进行,切片经甲苯胺蓝染色。接着切割50纳米超薄切片,并用 醋酸双氧铀进行对比染色。使用配备Megaview III数码相机和Radius 2.0软件(EMSIS,德国明斯特)的 透射电子显微镜采集电镜图像。通过拼图扫描成像量化锥体神经元胞体中线粒体长轴长度,采用单帧电镜图像采集CA1与CA3区锥体层的突触结构。使用Fiji软件(美国贝塞斯达NIH)进行形态计量学分析,通过突触后电子密度识别兴奋性突触。将矢状面直径介于20-80纳米的结构定义为突触小泡,与活性带接触的突触小泡被归类为锚定突触小泡。
¶ 统计分析
在每组实验中,首先使用Shapiro-Wilk检验对数据分布进行正态性检验。采用F检验评估各组数据间的方差齐性。符合正态分布的数据以均值 标准误表示;每个散点图同时包含个体数据点。根据具体情况采用非配对学生t检验、单因素方差分析或双因素方差分析结合事后多重比较检验。对于未通过正态性检验的数据集,按需使用Mann-Whitney U检验和秩方差分析结合Dunn多重比较检验。未采用统计方法预先确定样本量,但本研究样本量与领域内既往文献报道规模相当。所有统计分析均通过GraphPad9(Prism软件)完成。