¶ 靶向反义寡核苷酸治疗可挽救脊髓性肌萎缩类器官的发育异常
¶ 发表期刊:Natrue Communications
¶ 发表时间:2025年12月21
¶ 原文下载:41467_2025_article_67725.pdf
艾琳·法拉韦利1,15,18,林切蒂1,16,18,莫妮卡·坦巴洛2,18,伊莉莎·西姆廷2,丽莎·马佩利3,萨拉·曼奇内利2,4,马特奥·米奥托4,马法尔达·里祖蒂5,安德里亚·多奥5,恰拉·科迪格利里6,朱利亚·福罗蒂5,西莉亚·皮亚诺7,17,保罗·昆德弗兰科8,卢卡·卡兰德里埃洛5,P.科米1,5,埃尔韦齐亚·帕拉博斯奇2,埃莉奥诺拉·帕利3,比阿特丽斯5,埃吉迪奥·多奥9,Sreer Przedborski 10,11,12,13,莫妮卡·尼扎多5,西蒙娜·洛达托2,4和斯特凡尼亚·科蒂1,14
- 意大利米兰大学病理生理学与移植系(DEPT)神经科学部迪诺·费拉里中心
- 意大利人道主义大学生物医学科学系,米兰省皮耶韦埃马努埃莱
- 意大利帕维亚大学脑与行为科学系
- 意大利米兰省罗扎诺市人道主义临床与研究医院(IRCCS)
- 意大利米兰市卡格兰达波利多里科综合医院IRCCS基金会神经科学系神经科
- 意大利米兰市“罗密欧与恩里卡·因韦尔尼齐”国家分子遗传学研究所
- 意大利国家研究委员会米兰大学分部遗传与生物医学研究所,米兰省罗扎诺市
- 意大利米兰省罗扎诺市人道主义临床与研究医院(IRCCS)生物信息学组
- 意大利帕维亚市蒙迪诺基金会IRCCS数字神经科学中心
- 美国纽约州纽约市哥伦比亚大学运动神经元生物学与疾病研究中心
- 美国纽约州纽约市哥伦比亚大学病理学与细胞生物学系
- 美国纽约州纽约市哥伦比亚大学神经病学系
- 美国纽约州纽约市哥伦比亚大学神经科学系
- 意大利米兰市卡格兰达波利多里科综合医院IRCCS基金会神经肌肉与罕见病亚专科
- 现工作单位:美国马萨诸塞州剑桥市哈佛大学干细胞与再生生物学系
- 现工作单位:美国纽约州纽约市哥伦比亚大学系统生物学系
- 现工作单位:意大利米兰市人类技术园区
- 同等贡献作者:艾琳·法拉韦利、保拉·林凯蒂、莫妮卡·坦巴洛
通讯邮箱:simona.lodato@hunimed.eu;stefania.corti@unimi.it
脊髓性肌萎缩症(SMA)是一种由该基因发生突变引起的严重神经系统疾病,其特征是下运动神经元早发并退化。了解SMA早期的神经发育缺陷对于优化治疗干预措施至关重要。通过使用从多名1型SMA男性捐赠者体内生成的脊髓类器官和脑类器官,我们揭示了除运动神经元退化之外广泛存在的疾病机制。单细胞转录组学发现,从祖细胞到神经元,神经群体中普遍存在改变,这表明神经元分化程序的调节依赖于SMN,且出现了失调。多电极阵列(MEA)分析发现,脊髓类器官和脑类器官中均存在持续的过度兴奋,这证实了电特性改变是中枢神经系统范围内发病机制的一个特征。早期施用一种经过优化的反义寡核苷酸(ASO)可提高SMN水平,从而挽救不同遗传背景下脊髓类器官的形态和功能缺陷。重要的是,这种早期干预精确纠正了此处发现的、在神经元分化关键节点富集的靶标的异常剪接。我们的研究结果表明,早期发育缺陷是SMA发病机制的核心特征,及时的治疗干预可以预防这些缺陷,这为优化治疗策略提供了思路。
脊髓性肌萎缩症(SMA)是一种严重的遗传性神经系统疾病,仍是儿童死亡的主要遗传原因之一1,2。SMA由SMN1基因的常染色体隐性突变引起,导致全长SMN蛋白表达降低3。这会导致更脆弱的脊髓运动神经元(MNs)进行性退化,引起近端肌肉无力和萎缩,以及危及生命的早期呼吸衰竭4,5。SMA的表型谱非常广泛,包括从致命到轻度的不同临床类型,这些类型根据患者达到的运动里程碑进行分级6。
SMN蛋白在多种胎儿组织中表达,包括脑、脊髓、骨骼肌、心脏和肾脏;出生后其表达量显著下降,这表明SMN在胎儿期尤为重要7,8。来自人类SMA尸检的最新数据显示,子宫内运动神经元轴突发育异常与SMA患者出生后的变性相关9。除脊髓受累外,已有报道称SMA患者存在认知功能缺陷和脑神经病理学改变,但相关记录尚不完整10。驱动变性的早期事件及其对整个中枢神经系统(CNS)的影响在很大程度上仍不明确:在建立疾病模型时,考虑症状前因素至关重要。
最近,美国食品药品监督管理局(FDA)/欧洲药品管理局(EMA)批准了突破性的治疗策略来恢复运动神经元存活蛋白(SMN)的水平,这些策略目前已投入临床使用5,11。这些方法要么基于SMN1的cDNA替代,要么使用反义寡核苷酸(ASOs)或小分子来实现SMN2的剪接校正。SMN2是SMN1的同源基因,其产生的SMN蛋白截短且降解迅速,可作为恢复SMN水平的作用靶点12。这些药物极大地改变了脊髓性肌萎缩症(SMA)的临床预后,对于致命性至重度的1型SMA,能够通过阻止或预防运动功能障碍,将疾病转化为慢性状态。尽管越来越多的临床数据表明,已获批的治疗方法在慢性期也能带来益处,但有证据显示,对患者进行症状前治疗时,疗效最为显著5。尚未满足的治疗需求包括症状出现后的患者治疗5,以及对阻碍治疗 efficacy的因素的难以捉摸的理解。关键的是,即使是通过筛查确定的新生儿,其运动功能恢复也不完全,尤其是那些有两个SMN2拷贝的新生儿13。因此,更好地理解产前疾病阶段的SMA发病机制,对于优化当前治疗方法、提高效率、靶向生物分布以及制定新的临床策略至关重要。
传统的二维培养和动物模型只能部分重现人类运动神经元疾病的表型和遗传复杂性。相比之下,中枢神经系统的三维人类类器官14–16表现出高度的细胞多样性,使其能够模拟复杂的细胞间相互作用,同时模拟患者特异性的遗传背景。
在此,我们利用一种高效的方案来生成诱导多能干细胞(iPSC)衍生的脊髓类器官(SCOs),并揭示了与脊髓性肌萎缩症(SMA)早期临床相关的病理表型。我们检测到祖细胞行为的改变、早期神经元分化缺陷,这些甚至在细胞死亡的标志出现之前就已存在,并且还发现了除运动神经元(MNs)之外的不同神经元亚型的疾病易感性。
剪接机制的普遍改变与多种神经退行性疾病相关,最近有研究推测其与成人运动神经元疾病肌萎缩侧索硬化症(ALS)有关¹⁷。SMN蛋白在剪接机制中具有公认的作用,但其功能异常对发育过程的影响仍有待探索。利用脊髓类器官(SCOs),我们鉴定出在脊髓性肌萎缩症(SMA)中存在异常剪接的基因,以及那些通过SMN靶向治疗得到纠正的基因。这使我们能够研究在患者来源的脊髓类器官中,SMN的恢复如何重塑转录图谱。有趣的是,我们发现SMN缺乏会破坏参与神经分化的关键基因的剪接,这可能引发脊髓性肌萎缩症的发病机制。我们的研究结果证实了SMN在神经发育中的关键作用,以及其恢复在治疗策略中的重要意义。
¶ 结果
¶ SMN缺陷影响脊髓中的祖细胞和神经元
我们从健康对照者(CTRL,n=5)和1型脊髓性肌萎缩症(SMA,n=5)患者来源的诱导多能干细胞系中衍生出了人类脊髓类器官(SCOs)(补充图1和补充数据1)。脊髓发育是一个复杂的过程,既需要由Wnt和视黄酸(RA)介导的头尾轴特化,也需要由音猬因子(SHH)信号下游调控的背腹轴模式形成¹⁸,¹⁹。因此,我们在培养SCOs时加入了作用于这些通路的小分子,以在体外重现脊髓特化过程²⁰⁻²²(图1a)。在对照和SMA类器官中,这种处理都促进了渐进性的尾部神经化,这一点从分化早期(1个月)出现的神经祖细胞得到证实,这些祖细胞表达PAX6和NESTIN(图1b)。随后出现了神经元标志物DCX和MAP2,表明神经元分化正在进行(图1b、补充图2以及补充数据2和3)。随着分化的进行,我们观察到存在表达HOXB4和OLIG2的脊髓前体细胞,以及ISL1和MNX1阳性的运动神经元(图1b和补充图2)。在2个月时,我们还检测到了星形胶质细胞的存在(补充图2)。总之,这些数据表明,脊髓类器官能够重现脊髓神经发生的关键方面。
为探究驱动脊髓性肌萎缩症(SMA)发病机制的早期事件,我们通过单细胞RNA测序分析了2月龄对照(CTRL)和SMA脊髓类器官(SCOs)的组成(C1、C3、S1和S3系,图1c-e、补充图3及补充数据4)。考虑到技术变异性,我们对来自2个对照系和2个SMA系的单个类器官进行了测序(补充数据1),成功分析了共计27114个细胞。利用背侧和腹侧区域的特定标记基因集进行模块注释,识别出了不同类型的神经祖细胞(Prog,SOX2、SOX9),包括底板祖细胞(pFP,SHH)、分化中的神经元(Neuron,DCX)以及脊髓神经元区域的亚型(图1c-e及补充图3)。在这一阶段,我们还能识别出运动神经元前体(pMN,OLIG2)和有丝分裂后运动神经元(MN,PHOX2B、ISL1)的不同集群(图1c-e、补充图3d-g、补充图4及补充数据4)。与后神经管模式相关的特定HOX转录本组合的选择性表达,表明其获得了颈段脊髓的特性(补充图3d)。为了将对照类器官与人类胚胎及胎儿脊髓发育进行基准对比,我们使用了两个互补的转录组数据集。第一个是涵盖广泛发育阶段的单细胞RNA测序数据集23,这使我们能够确定与我们的类器官最相似的时间点,结果显示其与妊娠早期脊髓具有高度的转录相似性,在妊娠第8周达到峰值(补充图3e)。第二个数据集24是人类胎儿脊髓(妊娠4-8周)的高分辨率单细胞图谱,它能够精确绘制类器官的细胞组成。该分析表明,类器官与主要神经细胞类型(包括祖细胞和神经元)高度匹配,而与中胚层或内胚层谱系缺乏相似性(补充图3f、g)。综上所述,这些发现支持将脊髓类器官用作研究涉及多细胞的神经系统疾病(如SMA)的可靠体外模型。
为了研究在缺乏SMN蛋白的情况下,细胞组成和神经元类型的相对贡献是否发生改变,我们使用MiloPy框架进行了差异丰度测试(图1f、g)。结果显示,SMA类器官中特定的细胞亚型存在差异表达,其中运动神经元(MNs)在SMA基因型中显著减少。
我们进一步扩展了分析范围,纳入了不同基因型/品系之间的成对比较,以评估我们的研究结果在不同条件下的一致性。除了C1与S3的比较外,所有品系间的比较均显示,与对照组(CTRL)相比,脊髓性肌萎缩症(SMA)中的运动神经元(MN)数量显著减少。考虑到体外系统固有的变异性,这些结果仍然表明在此阶段运动神经元的数量显著减少(补充图5)。有趣的是,与运动神经元减少形成对比的是,脊髓性肌萎缩症类器官中的运动神经元前体细胞(pMN)以及其他神经祖细胞群均有所富集(图1f、g)。这些结果表明,生存运动神经元(SMN)的减少对神经多样性具有更广泛的影响,这会改变发育中的类器官中不同细胞类型的比例。
因此,我们研究了每个细胞簇中与SMN缺失相关的转录变化。为了考虑SMA类脊髓器官(SCOs)中细胞丰度的差异,我们对每个基因型细胞占比至少25%的细胞簇进行了差异基因表达分析(图1h和补充图6;绝对对数倍变化≥0.5,调整后p值<0.05)。在归属于运动神经元(MN)簇的细胞中,观察到失调基因的比例最大(对照组和SMA类脊髓器官之间有2031个差异表达基因,其中772个上调,1259个下调),而祖细胞是受影响第二大的细胞(补充数据5)。有趣的是,与祖细胞存活能力相关的特定基因(ASCL1、SOX4)表达下调,而参与凋亡信号传导的基因表达上调(图1i)。对祖细胞的基因本体分析(图1j)显示,SMN缺失对多个发育过程相关通路有广泛影响。总体而言,这些发现凸显了SMN缺陷对类脊髓器官的细胞组成和转录景观的深远影响。虽然脊髓运动神经元的显著减少是脊髓性肌萎缩症(SMA)的一个关键特征,但祖细胞丰度的改变和转录失调揭示了该疾病病理中一个同样关键的方面,即神经发育的更广泛破坏,这在对该疾病的研究中往往被忽视。
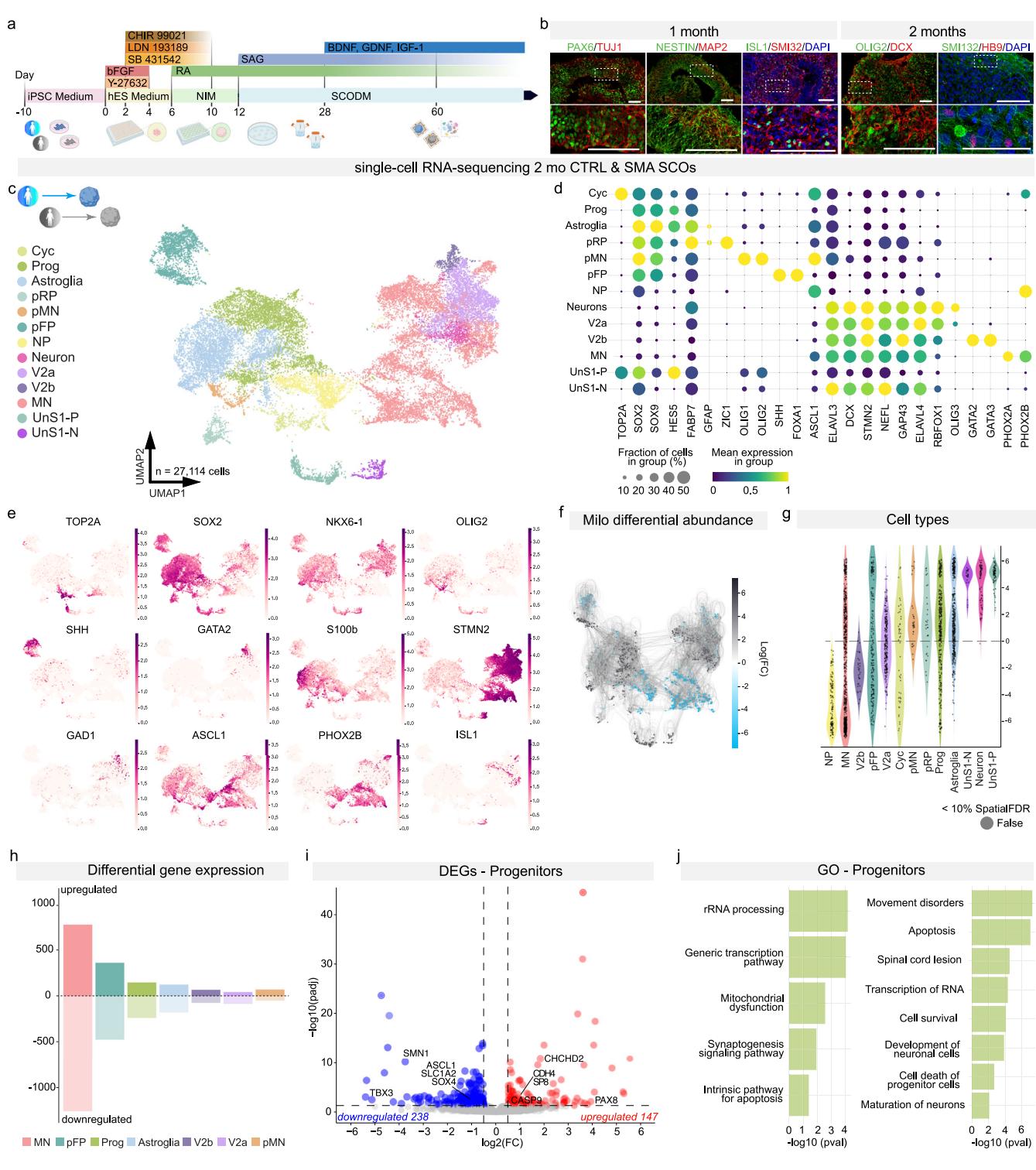
图1 | CTRL和SMA的脊髓类器官(SCOs)显示出不同亚型的脊髓细胞群。a 人类脊髓类器官(SCO)生成示意图。使用BioRender制作。
Rinchetti, P.(2025)https://BioRender.com/l7ay5hq。b 1个月和2个月时SCO中发育标志物和神经元标志物的免疫染色结果。SCO对神经发育标志物(包括PAX6、NESTIN和OLIG2,绿色)以及神经元标志物(如ISL1(绿色)、TUJ1、MAP2、SMI32和DCX,红色)呈阳性染色。此外,运动神经元通过HB9(红色)的阳性表达来指示。细胞核用DAPI(蓝色)染色。比例尺:100μm。代表性图像。每个批次至少对3个不同的类器官进行独立免疫染色,至少包含3个CTRL系和3个SMA系。c 来自C1、C3、S1和S3系的2个月(mo)时的CTRL和SMA单细胞RNA测序结果。无监督UMAP将SCO分为神经祖细胞( Cycling细胞(Cyc)、祖细胞(Prog)、星形胶质细胞、顶板祖细胞(pRP)、运动神经元祖细胞(pMN)、底板祖细胞(pFP)、神经源性祖细胞(NP))、分化中的神经元细胞(Neuron)、几种腹侧神经元亚型(V2a、V2b)、运动神经元(MN),以及一个独特的未匹配SMA细胞簇,该簇特别源自SMA S1系(未匹配S1祖细胞(UnS1-P)、未匹配S1神经元(UnS1-N))。d 点图显示与细胞分化相关的代表性基因的簇特异性表达;点的大小对应于簇中表达每个基因的细胞百分比,颜色对应于平均表达水平(从紫色到黄色,代表从低到高)。e UMAP特征图显示增殖因子(TOP2A和SOX2)、祖细胞标志物(NKX6-1、OLIG2、SHH、GATA2)和神经元分化标志物(STMN2、GAD1、PHOX2B和ISL1)的选定代表性基因的对数归一化计数。f Milo差异丰度测试结果的邻域抽象图。灰色表示SMA富集,蓝色表示CTRL富集。g 小提琴图显示分配给细胞类型标签的差异丰度邻域的对数倍变化分布。h 不同簇中失调基因的数量。i SMA SCO与CTRL相比,祖细胞簇中差异基因表达(DEG)的火山图。每个基因的log2倍变化值相对于−log10 q值作图。红色表示SMA中上调的基因,蓝色表示SMA中下调的基因(绝对对数倍变化≥0.2,最小百分比=0.25,调整后的q值<0.05,阈值用虚线标出)。j 使用QIAGEN Ingenuity Pathway Analysis(IPA)软件对祖细胞簇中上调和下调基因进行的选定基因本体论(GO)分析(采用Benjamini-Hochberg校正的Fisher检验)。
¶ 脊髓性肌萎缩症祖细胞缺陷先于运动神经元损伤
为了在细胞水平上纵向捕捉分化进程表型,我们进行了高内涵图像分析(图2a-d)。值得注意的是,在SMA类脊髓器官(SCOs)中观察到了异常的分化轨迹。对照组(CTRL)的SCOs在1至2个月内,SOX2阳性神经祖细胞明显减少,而有丝分裂后神经元标志物TUJ1则增加;而SMA的SCOs中,SOX2的减少不太明显,且TUJ1阳性细胞的比例没有增加(图2a-d;每个类器官中SOX2阳性细胞在1个月 vs 2个月的百分比:CTRL为47.57%±11.94 vs 20.40%±13.85,p<0.0001;SMA为38.79%±14.42 vs 25.44%±13.91,p=0.0148;每个类器官中TUJ1阳性细胞在1个月 vs 2个月的百分比:CTRL为13.52%±4.788 vs 28.42%±12.35,p=0.0002;SMA为21.49%±14.28 vs 13.84%±6.759,p=0.0764)。相应地,对照组在第30天时SOX2/TUJ1比值显著较高(p=0.0444),但到第60天时显著下降,这表明随着时间的推移,细胞从祖细胞向分化的神经元转变,与预期的生理轨迹一致。然而,在SMA情况下未观察到这种模式(图2e)。
由于类脊髓器官(SCOs)能重现脊髓发育的早期阶段,且我们的数据表明脊髓性肌萎缩症(SMA)中细胞死亡的生理过程可能会增强,因此我们对对照组(CTRL)和SMA的类脊髓器官进行了程序性细胞死亡的纵向检测。与对照组类脊髓器官相比,SMA类脊髓器官的凋亡动态确实存在差异,在2个月时凋亡显著增强(每个类器官中凋亡细胞核的百分比在1个月 vs 2个月时:对照组为11.30%±2.512 vs 7.369%±5.125,p=0.3786;SMA为4.245%±2.372 vs 23.02%±11.34,p<0.0001;补充图7a、b)。基于SMA和对照组类脊髓器官之间的差异表达基因(DEG),对凋亡相关生物学功能进行的富集分析显示,这些过程在SMA的运动前神经元(pMNs)和运动神经元(MNs)中主要呈上调状态(补充图7c)。
细胞死亡率的差异或许至少能部分解释为何MiloPy捕获的SMA类脊髓器官(SCOs)中运动神经元(MN)数量减少(图1f、g),而细胞类型组成的改变则表明SMA组织的早期发育程序发生了变化。
为了研究从祖细胞到神经元状态的发育动态差异,我们使用CytoTRACE(通过CellRank框架实现)对细胞进行了伪时间排序(图2e)。该方法基于转录复杂性推断细胞分化状态,重要的是,它不依赖于根细胞的选择,能够在无需对谱系起始点进行预先假设的情况下实现无偏的伪时间排序。与人类胎儿体内发育情况一致,CTRL类器官呈现出从祖细胞群到分化神经元细胞类型的连续进展(图2e、f)。相比之下,SMA SCOs在整体上显著向发育成熟度较低的方向偏移(柯尔莫哥洛夫-斯米尔诺夫(KS)检验,p = 2.16e-08),这在所有基因型中均有体现(补充图9)。细胞类型特异性分析进一步显示,伪时间评分分布存在显著的统计学差异(图2g)。
总之,这些数据支持了我们之前的研究结果,即脊髓性肌萎缩症(SMA)神经发育存在早期异常,表现为细胞停留在中间发育阶段,无法继续成熟,这可能反过来影响它们在新生神经回路中的存活能力。
¶ SMA运动神经元的发育轨迹受阻
为了研究运动神经元(MN)分化过程中的基因表达变化,我们对数据集进行了划分,专门聚焦于MN谱系,包括pMN、早期运动神经元和成熟运动神经元(图3及补充图8)。我们确定了躯体运动神经元的主要类别,几乎没有证据表明存在明显的内脏运动神经元群体,因为内脏标志物要么不存在,要么与骨骼肌运动神经元标志物共表达(补充图8b)。通过对MN谱系亚群进行扩散映射,我们观察到8个不同的转录簇(补充图8a),基于PHOX2B与ISL1/MNX1的表达水平,这些转录簇对应于三种不同的MN亚型(补充图8b-e)。尽管在CTRL基因型中MN细胞的总体丰度更高(图1f、g),但MN-1 ISL1⁺/MNX1⁺亚群在SMA基因型中优先富集。
接下来,我们探究了运动神经元分区内的发育路径,从而能够绘制从pMN到成熟运动神经元的神经发生进程(图3a-c)。在CTRL类脑器官中,从pMN到运动神经元的神经发生轨迹遵循了分化过程中预期的顺序进展(图3b、c)。SMA类脑器官的运动神经元分化轨迹则呈现出不同的情况。与CTRL样本中观察到的连续进展不同,SMA中的分化轨迹似乎呈极化状态。这种极化的特征是:在轨迹起始处,祖细胞状态的细胞高度集中;中间状态的细胞大量积累;而在轨迹末端,运动神经元显著富集,且主要集中在单一分支中(图3c)。重要的是,对数据集按单个细胞系进行分析时也得到了一致的结果,这表明尽管不同细胞系中运动神经元的相对丰度存在差异,但这种潜在的发育改变是所有细胞系共有的特征(补充图9)。这提示,SMN蛋白的缺乏可能会干扰运动神经元分化过程中细胞命运的逐步获得,导致祖细胞与终末分化神经元之间的失衡。
特定转录因子(TFs)之间的相互作用对于脊髓中正确发育程序的执行至关重要26;细胞类型特异性转录因子是基因调控网络的核心枢纽,其表达的改变与细胞特性获得和维持的下游缺陷相关18。为了研究与脊髓性肌萎缩症(SMA)相关的早期发育缺陷是否由转录因子活性的变化所驱动,我们使用pySCENIC(单细胞调控网络推断与聚类)算法27作为工具,来筛选CTRL和SMA脊髓类器官(SCOs)发育轨迹中的关键转录因子(图3d、e)。有趣的是,我们发现几个已知调控人类脊髓分子发育关键方面的转录因子(即SOX2、SOX9、OLIG2、NKX6-1)在SMA和CTRL的脊髓类器官中,沿着SOX2、SOX9、OLIG2、NKX6-1的不同状态轨迹表达(图3d、e),这表明在缺乏SMN的情况下,不同细胞状态间转录程序的活性存在重要差异。我们还鉴定出了组蛋白修饰因子和染色质结构调节因子(CHD2、HDAC2、KDM5A),它们可能是人类脊髓发育的重要决定因素,并可能影响SMA的病程。
为了确定发育性转录因子(TF)的表达是否暗示了不同的基因调控程序,我们计算了调控子活性。我们观察到,对于运动神经元(MN)分化至关重要的转录因子的共有调控子(例如SOX2、PHOX2B、CHD2)在脊髓性肌萎缩症(SMA)中呈现出活性特征的改变,其转录程序的活性持续存在,这与祖细胞状态的长期维持相关(图3d、e)。总之,这些分析支持以下观点:脊髓性肌萎缩症类脊髓器官(SMA SCOs)不仅无法正常分化为成熟的运动神经元,而且其神经元发育轨迹及相关的转录调控从早期祖细胞状态就发生了显著改变。这可能会影响运动神经元的存活,并对整个脊髓神经元的分化产生影响。
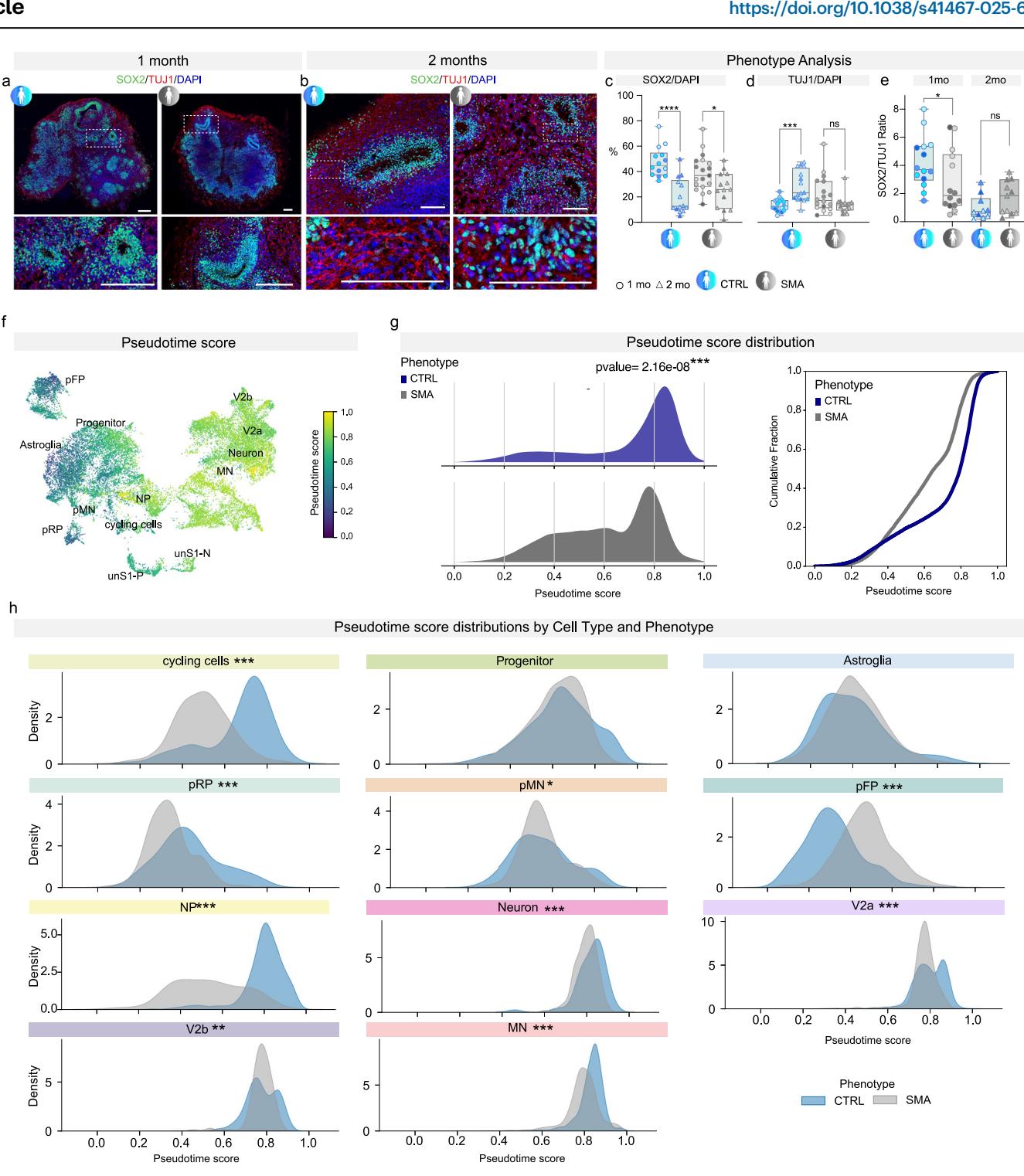
图2 | SMA类脊髓器官(SCOs)的神经发育疾病特征。a、b 对照组(CTRL)和SMA类脊髓器官在1个月和2个月时SOX2(绿色)和TUJ1(红色)染色的代表性图像。比例尺:200 μm。细胞核用DAPI(蓝色)染色。c SOX2阳性细胞的定量分析(占DAPI染色显示细胞的百分比),分别在1个月(圆圈)和2个月(三角形)(双因素方差分析,Sidak多重比较检验:对照组和SMA的p值分别为<0.0001和0.0148。来自30天和60天对照组类脊髓器官的样本数n=28:n=4 C1、n=4 C2、n=3 C4、n=3 C5;n=8 C1、n=3 C4、n=3 C5;来自30天和60天SMA类脊髓器官的样本数n=33:n=4 S1、n=7 S2、n=6 S4、n=2 S5;n=5 S2、n=4 S3、n=5 S4)。箱线图显示系列中的最小值和最大值,以及中位数和第25-75百分位数。d 在对照组中,TUJ1的表达随时间增加,而在SMA类脊髓器官中,TUJ1的表达有下降趋势(双因素方差分析,Sidak多重比较检验:对照组和SMA的p值分别为0.0002和0.0764。来自30天和60天对照组类脊髓器官的样本数n=33:n=4 C1、n=5 C2、n=3 C3、n=3 C4、n=3 C5;n=4 C1、n=3 C2、n=3 C3、n=3 C4、n=2 C5;来自30天和60天SMA类脊髓器官的样本数n=33:n=4 S1、n=2 S2、n=4 S3、n=6 S4、n=2 S5;n=4 S1、n=3 S2、n=3 S3、n=5 S4)。e 堆叠条形图显示对照组和SMA类脊髓器官在1个月和2个月时SOX2与TUJ1的比例(Mann-Whitney检验:1个月和2个月时对照组与SMA相比,p值分别为0.0444和0.1734。来自30天和60天对照组类脊髓器官的样本数n=24:n=4 C1、n=4 C2、n=3 C4、n=3 C5;n=5 C1、n=3 C4、n=2 C5;来自1个月和2个月SMA类脊髓器官的样本数n=25:n=4 S1、n=2 S2、n=6 S4、n=2 S5;n=3 S2、n=3 S3、n=5 S4)。f UMAP图显示按伪时间分数着色的细胞,并标注主要细胞类型。g 左图:对照组(蓝色)和SMA(灰色)细胞中伪时间分数分布的密度图(双侧Kolmogorov-Smirnov(KS)检验,p=2.16e-08)。右图:对照组和SMA细胞中伪时间分数的累积分布图。h 各细胞类型伪时间分数的密度图,比较对照组(蓝色)和SMA(灰色)。使用双侧KS检验评估伪时间分布的显著差异。星号表示显著性水平:*p<0.05,**p<0.01,***p<0.001,****p<0.0001。源数据作为源数据文件提供。
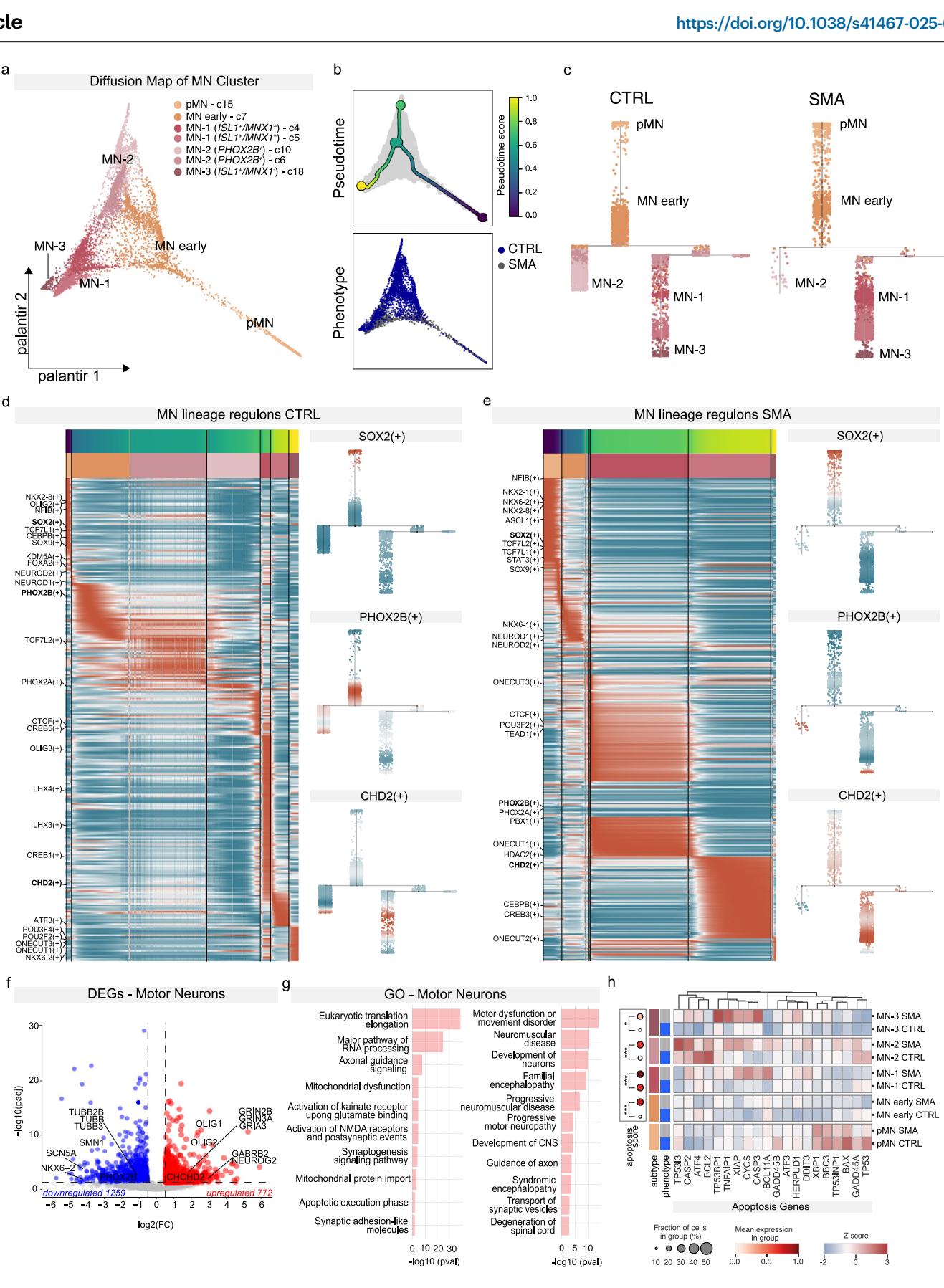
图3 | SMA SCOs显示疾病的分子特征。a 运动神经元(MN)集群的扩散图(DM),按亚集群特征着色。b 上图:按拟时间评分着色的扩散图。下图:按样本表型(CTRL,SMA)着色的扩散图。c CTRL和SMA样本的运动神经元在拟时间轨迹上的投影。细胞按亚集群特征着色,以显示其沿轨迹的分布。d、e 热图显示与CTRL(d)和SMA(e)轨迹相关的调节子活性趋势和拟时间,按每个亚集群特征分箱。CTRL和SMA样本中SOX2、PHOX2B和CHD2的调节子活性评分。调节子活性高的以红色表示,活性低的以蓝色表示。f 与CTRL相比,SMA SCOs中运动神经元集群的差异基因表达(DEG)火山图。每个基因的log2倍变化值与q值的−log10作图。红色表示SMA中上调的基因,蓝色表示SMA中下调的基因(绝对log倍变化≥0.2,最小百分比=0.25,校正q值<0.05,阈值以虚线绘制)。g 使用QIAGEN Ingenuity通路分析(IPA)软件对运动神经元中上调和下调基因进行的选定基因本体论(GO)分析(采用Benjamini–Hochberg校正的Fisher检验)。h 左图:显示凋亡模块评分表达的点图。右图:不同运动神经元亚集群和表型中凋亡相关基因表达的热图。
然后,我们研究了脊髓性肌萎缩症(SMA)与对照组(CTRL)运动神经元(MNs)的转录图谱(图3f、g)。SMA运动神经元中,与神经元生长和突触成熟相关的基因(TUBB3、TUBB、SCN5A)表现出下调。值得注意的是,一小部分基因在SMA类脊髓器官(SCOs)的几乎所有细胞类型中都存在一致的表达失调(补充数据5)。其中包括CHCHD2,这是一种参与氧化磷酸化的线粒体蛋白,先前研究表明,在其他神经退行性疾病中,其上调是对氧化应激的一种代偿性保护反应²⁸,²⁹。有趣的是,已有研究显示CHCHD2存在可变剪接现象。我们通过定量聚合酶链反应(qPCR)验证,在来自两个独立的对照组和SMA细胞系的各个类脊髓器官中,该基因均重现了相同的表达失调趋势(补充图10a),而在多株未分化的SMA诱导多能干细胞(iPSCs)中未观察到差异(补充图10b)。总体而言,SMA类脊髓器官在各种运动神经元成熟状态下均表现出与疾病相关的分子特征,导致多个分子程序发生整体改变。基因本体论分析确定了与代谢过程、应激反应和脊髓疾病相关的富集类别(图3g)。事实上,在来自解离的类脊髓器官的SMA神经元中,检测到神经突起显著减少(通过重神经丝NEF-H/SMI32染色识别,这是一种在运动神经元中富集的标记基因)(对照组为59.17±12.30μm,SMA组为34.62±8.34μm,p<0.0001;补充图8f、g)。最后,我们通过计算对照组和SMA条件下每个运动神经元分区中细胞凋亡的模块分数,研究了与细胞死亡相关的分子程序(图3h)。我们发现,在所有亚组的SMA运动神经元中,与细胞死亡相关的分子程序显著富集。这表明,尽管在此时点SMA中某些运动神经元亚型的占比更高,但它们不仅表现出发育轨迹的失调,而且已经启动了与运动神经元丢失相关的退行性级联反应。
SMA脊髓类器官中的病理性活动模式 先前已有研究报道,在2D培养物³⁰和SMA症状前小鼠模型³¹中,脊髓运动神经元(MNs)的电生理活动存在改变。与其他运动退行性疾病³²一样,这种异常活动可能会促成SMA的人类疾病表型。然而,目前尚不清楚这种功能障碍在疾病进展中出现的时间有多早,以及它在中枢神经系统(CNS)各部分之间的差异有多大。我们利用高密度多电极阵列(HD-MEA)技术来研究源自SMA患者的脊髓类器官(SCOs)的功能(图4a-c)。对2个月大的对照(CTRL)和SMA脊髓类器官的基础记录显示,它们存在自发性尖峰活动,且SMA脊髓类器官的频率显著更低(对照:1.956±0.5354 Hz,SMA:1.355±0.6807 Hz,p=0.0127;图4b、c)。此外,在谷氨酸能刺激后,SMA脊髓类器官的放电频率增加幅度显著大于对照脊髓类器官,这表明存在具有过度兴奋性特征的早期功能障碍(对照:44.06%±20.03,SMA:91.03%±28.42,p<0.0001;图4b、c)。
值得注意的是,差异表达基因(DEG)分析显示,在不同的细胞簇中,不同神经元群体中存在一组与突触通道相关的失调基因,这表明其对突触功能有整体影响(补充图11a)。为了排除γ-氨基丁酸(GABA)能传递的变化,我们在基础水平上施用gabazine和士的宁³³以阻断GABA-A受体和甘氨酸受体,测试了自发放电频率。正如预期的那样,在对照(CTRL)类器官中观察到自发放电频率增加(CTRL 44.21%±5.626),在脊髓性肌萎缩症(SMA)脊髓类器官(SCOs)中也检测到类似的情况(SMA 43.75%±16.4,p=0.9521;补充图11b-d)。这种反应表明类器官回路中存在活跃的神经元抑制机制(CTRL 43.71%±36.38 vs. SMA 35.72%±19.11,p=0.5455)。GAD65染色证实,在对照和SMA脊髓类器官中均存在抑制性中间神经元(补充图11b下半部分),单细胞分子数据也突出显示了这一点。这些结果支持了以下假设:所观察到的SMA类器官对谷氨酸的过度兴奋反应并不意味着抑制性传递减少,更可能是由于谷氨酸能传递成熟过程中的改变所致。
总的来说,我们发现SCOs表现出基础放电活动,能够对外源性刺激做出反应,并且它们会形成功能性回路。此外,SMA的SCOs具有独特的电生理特性,包括谷氨酸敏感性增强,这可能导致过度兴奋——这是许多神经退行性疾病的共同特征。
¶ SMA脑类器官中的病理性活动模式
传统上,脊髓性肌萎缩症(SMA)被认为是一种累及脊髓运动神经元(MN)的疾病,然而,在最严重的SMA病例中已有脑受累的相关报道¹⁰。这一特征是与SMA的发病机制直接相关,还是与脊髓退行性变逆行扩散至大脑间接相关,目前仍存在争议。为了探究这些可能性,我们采用改良方案,从SMA患者和健康人的诱导多能干细胞(iPSC)系中生成3D脑类器官¹⁴(图4d、e及补充图12),排除了脊髓退行性变可能产生的继发影响。对照组(CTRL)和SMA组的脑类器官均呈现复杂的形态,其异质性区域包含神经祖细胞(SOX2+),这些细胞主要位于神经玫瑰花结内,同时还包含神经元(TUJ1+;图4f)。我们在RNA和蛋白质水平上对CTRL组和SMA组脑类器官中SOX2阳性祖细胞的数量进行了量化,发现在2个月时未检测到显著差异(CTRL组为57.45±19.10,SMA组为67.02±11.54,p=0.2449;图4g)。同时,我们也未检测到NEF-H/SMI32的表达存在任何显著差异(补充图12c)。在2个月时对神经胶质细胞群(GFAP+)、皮质神经元(CTIP2+和SATB2+)以及通用神经元标志物(DCX和MAP2)进行染色,结果显示存在多种细胞类型(补充图12a、b)。与脊髓类器官(SCOs)一样,SMA重编程的多能干细胞可以分化为脑组织,且脑类器官能够重现大脑发育的关键早期步骤。
有趣的是,大脑类器官表现出与SMA脊髓类器官相似的异常电生理活动。高密度微电极阵列(HD-MEA)分析(图4h、i)显示,与对照组(CTRL)相比,SMA大脑类器官的基础放电频率显著更低(对照组为2.206±0.6826 Hz,SMA组为0.8497±0.1629 Hz,p<0.0001;图4i)。与谷氨酸能刺激后观察到的脊髓类器官反应一致,SMA大脑类器官的放电频率增加幅度显著高于对照组类器官(SMA组为115.3%±36.20,对照组为39.11%±13.21,p<0.0001;图4i)。此外,还检测到存在利用GABA-A(可能还有甘氨酸)受体的活跃抑制性神经元回路(补充图12d、e)。因此,我们的结果显示SMA大脑类器官存在病理特征,表明该疾病直接累及中枢神经系统(不仅仅是脊髓),并揭示在临床治疗环境中需要考虑更多靶点,以评估治疗的疗效和长期反应。
¶ 优化的SMN靶向方法可有效恢复细胞和功能改变
在已获批的脊髓性肌萎缩症(SMA)治疗方法中,诺西那生钠基于一种作用于SMN2的反义寡核苷酸(ASO_10-27),可提高SMN蛋白的整体水平。为了确定SMA类器官是否是测试潜在治疗方法有效性的合适模型,以及提高SMN蛋白水平是否能纠正特定的分子疾病特征,我们测试了一种最近经过验证、具有优化吗啉代化学结构的SMN2反义寡核苷酸(MO-10-34³⁴,³⁵),该反义寡核苷酸与富含精氨酸的细胞穿透肽(r6-MO)偶联。我们最近证实,r6-MO能提高动物模型中药物的生物分布和疗效³⁶。
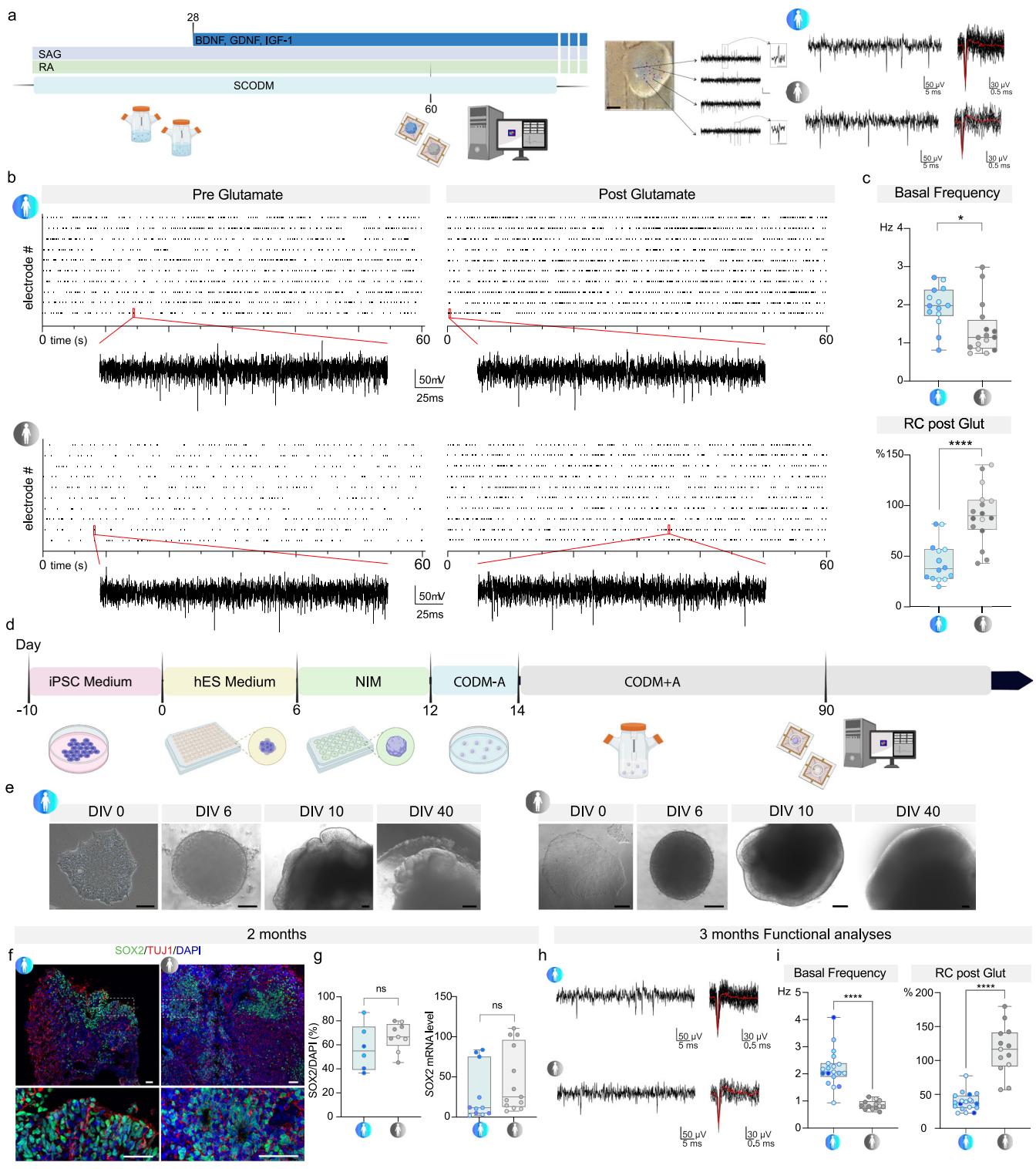
图4 | SMA脊髓类器官(SCOs)和大脑类器官(CrOs)的电生理活动发生改变,具有过度兴奋性特征。a MEA分析时间线示意图以及高密度MEA芯片上SCO的代表性图像,记录轨迹显示基础活动。比例尺:500 μm。示意图部分元素通过BioRender制作。Rinchetti, P.(2025)https://BioRender.com/0o9jsh1。b 对照组(CTRL)和SMA SCOs在谷氨酸处理前后的光栅图。c 上图:对照组与SMA SCOs的基础放电活动量化(非配对t检验,p = 0.0124,n = 29个类器官,其中对照组为n = 5 C1、n = 4 C3、n = 5 C4;SMA组为n = 1 S1、n = 4 S2、n = 7 S4、n = 4 S5)。下图:对照组与SMA SCOs在谷氨酸(glut)灌流后放电频率增加的相对变化(RC)(非配对t检验,p < 0.0001,n = 30个类器官,其中对照组为n = 5 C1、n = 4 C3、n = 5 C4;SMA组为n = 1 S1、n = 4 S2、n = 7 S4、n = 4 S5)。箱线图显示系列中的最小值、最大值以及中位数和第25-75百分位数。d 人类CrOs生成示意图。通过BioRender制作。Rinchetti, P.(2025)https://BioRender.com/33yw3mr。e 对照组和SMA CrOs分化过程的代表性明场图像。比例尺=100 μm。f 2个月时对照组和SMA CrOs中SOX2(绿色)和TUJ1(红色)染色的代表性图像。比例尺:200 μm。细胞核用DAPI(蓝色)染色。g 左图:2个月时CrOs中SOX2阳性细胞核百分比的量化及mRNA水平分析。在这两项分析中,对照组和SMA CrOs的SOX2表达无差异(左图:非配对t检验,p = 0.2449;n = 15个类器官,其中对照组为n = 3 C3、n = 3 C5;SMA组为n = 5 S1、n = 4 S3)。右图:非配对t检验,p = 0.2524;n = 24个类器官:对照组为n = 2 C1、n = 2 C3、n = 1 C4、n = 3 C5、n = 3 C6;SMA组为n = 3 S1、n = 3 S2、n = 7 S3)。箱线图显示系列中的最小值、最大值以及中位数和第25-75百分位数。h 对照组和SMA CrOs(3个月)基础记录的代表性轨迹。i 左图:SMA CrOs的基础放电频率低于对照组(非配对t检验,p < 0.0001,n = 32个类器官,其中对照组为n = 8 C1、n = 3 C2、n = 4 C3、n = 4 C6;SMA组为n = 2 S1、n = 5 S3、n = 6 S4)。右图:谷氨酸(glut)灌流后,SMA CrOs的放电频率相对增加大于对照组CrOs(非配对t检验,p < 0.0001,n = 32个类器官,其中对照组为n = 8 C1、n = 3 C2、n = 4 C3、n = 4 C6;SMA组为n = 2 S1、n = 5 S3、n = 6 S4)。箱线图显示系列中的最小值、最大值以及中位数和第25-75百分位数。原始数据见原始数据文件。
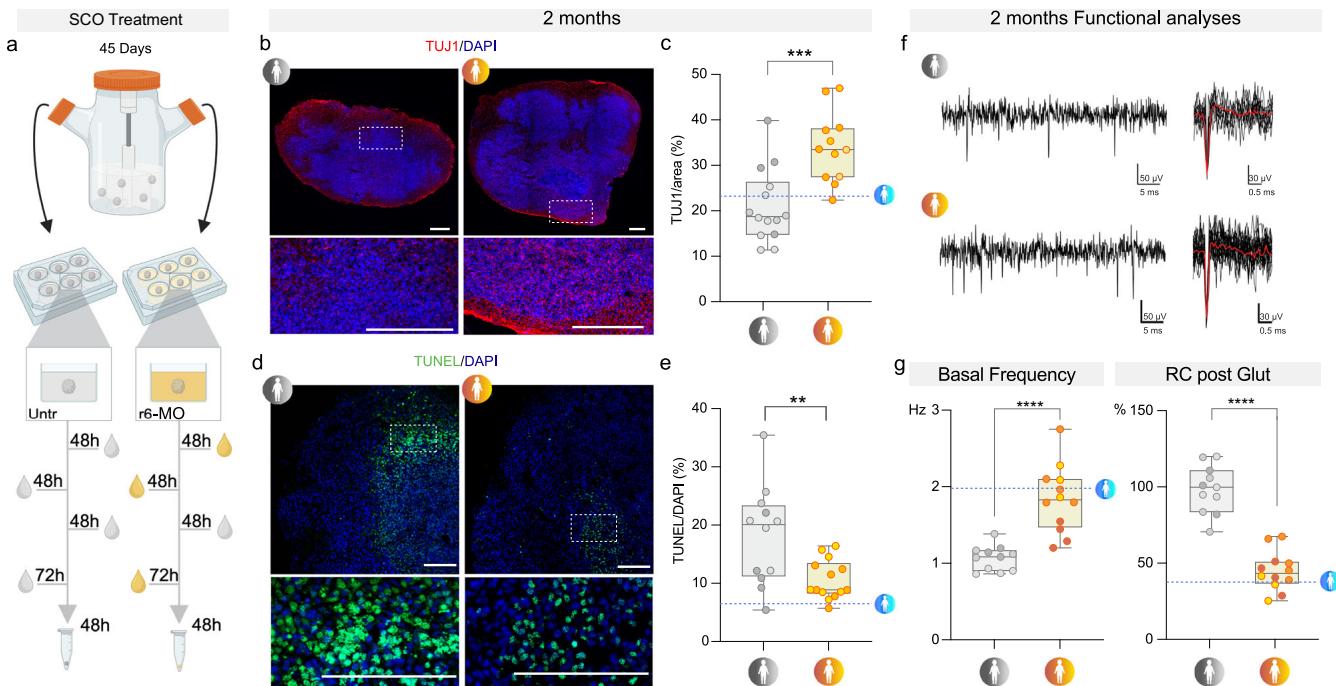
图5 | SMA类器官表现出特定的细胞特征,这些特征可通过r6-吗啉代得到挽救。a r6-吗啉代(r6-MO)处理的示意图。SMA SCOs共处理3次,每次48小时:T1、48小时后的T2、更换不含该化合物的培养基72小时后的T3。使用BioRender制作。Rinchetti, P.(2025)https://BioRender.com/cd9gra2。b 2个月时未处理(Untr)和r6-MO处理的SMA SCOs的TUJ1染色代表性图像。比例尺:300 μm。细胞核用DAPI(蓝色)染色。c r6-MO处理的SCOs中TUJ1的表达(强度/面积)高于未处理的SCOs(非配对t检验,p=0.0003,n=26个类器官,其中未处理组:n=7 S1、n=3 S2、n=4 S3;处理组:n=7 S1、n=3 S2、n=2 S3)。箱线图显示了系列中的最小值、最大值以及中位数和第25-75百分位数。d 2个月时未处理和r6-MO处理的SMA SCOs的TUNEL染色代表性图像。比例尺:300 μm。细胞核用DAPI(蓝色)染色。r6-MO处理的SCOs中TUNEL阳性细胞的数量少于未处理的SCOs(非配对t检验,p=0.0050,n=26个类器官,其中未处理组:n=4 S1、n=3 S2、n=5 S3;处理组:n=4 S1、n=3 S2、n=7 S3)。箱线图显示了系列中的最小值、最大值以及中位数和第25-75百分位数。f 2个月时未处理和r6-MO处理的SMA SCOs的基础记录代表性轨迹。g 左图:未处理组与r6-MO处理组SCOs的基础放电活动量化(非配对t检验,p<0.0001,n=23个类器官,其中未处理组:n=4 S1、n=4 S2、n=3 S3;处理组:n=4 S1、n=5 S2、n=3 S3)。右图:谷氨酸灌流(glut)后,与未处理组相比,r6-MO处理组SCOs放电频率降低的相对变化(RC)(非配对t检验,p<0.0001,n=23个类器官,其中未处理组:n=4 S1、n=4 S2、n=3 S3;处理组:n=4 S1、n=5 S2、n=3 S3)。箱线图显示了系列中的最小值、最大值以及中位数和第25-75百分位数。原始数据作为原始数据文件提供。
首先,我们研究了未偶联的MO-10-34和r6-MO是否能提高脊髓性肌萎缩症(SMA)类脊髓器官(SCOs)中SMN蛋白的水平。在第45天,我们用未偶联(裸)MO或r6-MO(2.4纳摩尔),或它们各自的定制乱序序列处理SMA类脊髓器官,持续48小时。这种处理以递增剂量(最高达4.8纳摩尔)重复了三次(图5a和补充图13a)。SMN蛋白减少是SMA病理的一个标志(补充图13b)。因此,我们首先评估了我们的处理是否能同时提高SMN蛋白和mRNA的水平。在较晚的时间点,与未处理的对照组相比,r6-MO处理的类器官裂解物中观察到SMN蛋白表达显著增加(补充图13c)。在类器官的外周和中央核心都检测到了生物素标记的r6-MO(补充图13d),表明其有效且广泛的组织分布。与此一致的是,在所有处理的细胞系(S1、S2和S3)中,与各自的乱序对照组相比,用r6-MO或MO-10-34(裸吗啉代)处理均导致SMN mRNA水平显著上调(补充图13e、f)。为了专门检测SMN1和SMN2亚型的表达,我们在对照(CTRL)、未处理的SMA、乱序处理的SMA和r6-MO处理的SMA细胞系中进行了限制性片段长度多态性(RFLP)分析。该分析证实,r6-MO处理显著降低了截短的SMN2 Δ7亚型的水平,而促进了全长SMN2转录本的表达(补充图13g)。
值得注意的是,r6-MO处理完全恢复了分化表型(TUJ1阳性细胞百分比:SMA组为20.99%±8.044,r6-MO组为33.93%±7.648,p=0.0003;图5b、c)。在第45天施用r6-MO也能显著阻止细胞死亡(2个月时每个类器官中凋亡细胞核的百分比:未处理组为18.16%±8.443,r6-MO组为10.58%±3.37,p=0.005;图5d、e),这支持了早期干预可以阻止异常细胞过程、积极改变疾病进程的观点。
值得注意的是,在功能层面上,与未处理的SMA类脊髓器官(SCOs)相比,r6-MO处理显著增强了基础活动(r6-MO组为1.843±0.4396 Hz,未处理组为1.067±0.1623 Hz,p<0.0001;图5f、g),同时对谷氨酸能刺激的反应降低(频率增加百分比:r6-MO组为44.76%±12.81%,未处理组为98.31%±15.56%,p<0.0001;图5f、g)。
这些研究结果表明,在多种遗传背景下,早期特定的SMN增加疗法可以挽救基本的细胞表型和功能表型。综上所述,这些数据为优化反义寡核苷酸(ASOs)的价值及其对脊髓性肌萎缩症(SMA)病理早期治疗的影响提供了见解。
¶ 增加SMN的疗法可纠正SMA的剪接缺陷
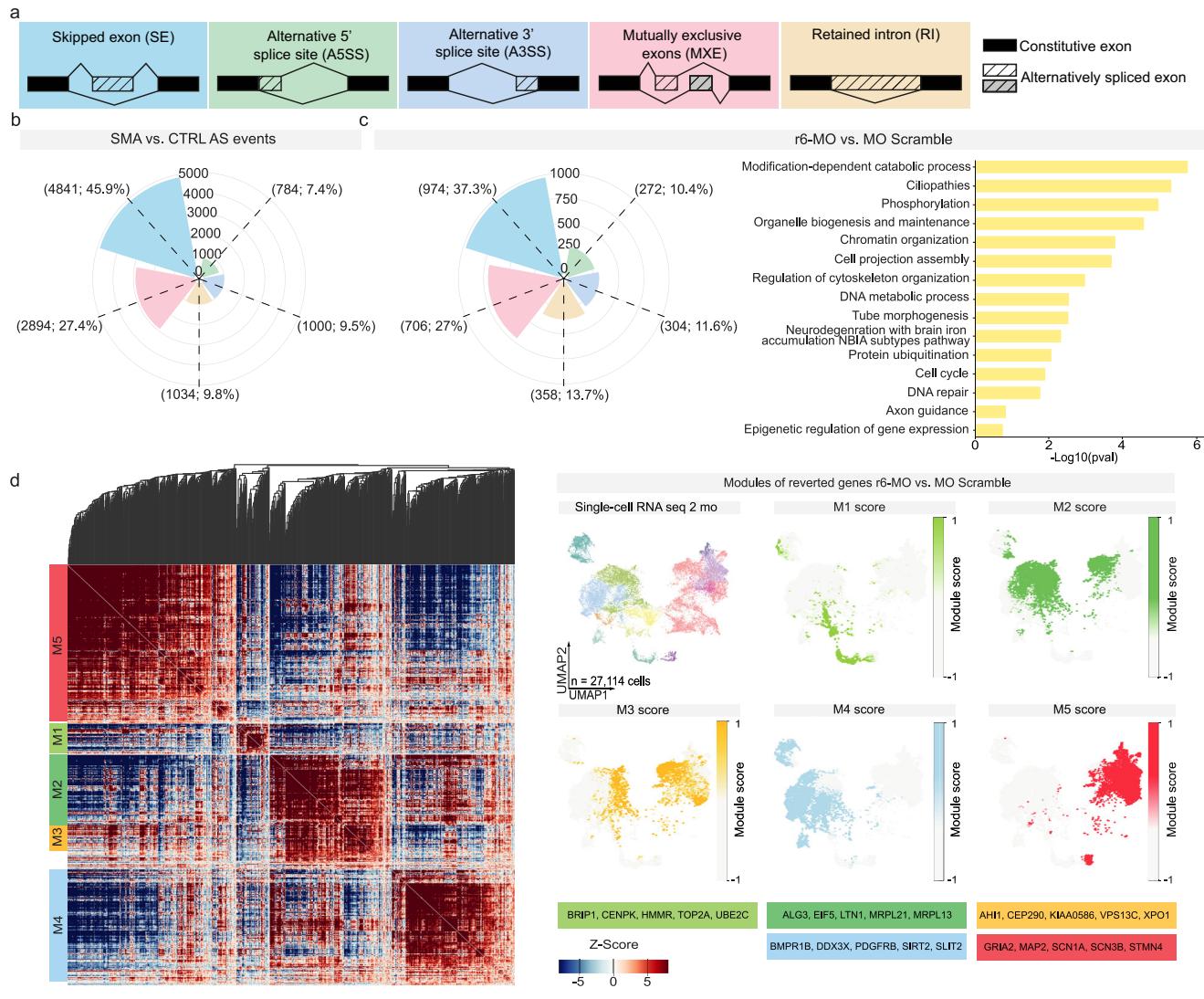
疾病修饰疗法目前正在改变脊髓性肌萎缩症(SMA)的自然病程,但SMN靶向疗法所影响的特定分子通路,以及这些通路在发育过程中对不同细胞类型的影响尚未完全阐明。我们利用自己的平台剖析了负责挽救细胞和功能表型的分子驱动因素。SMN蛋白在剪接机制中具有公认的关键作用³⁷。因此,通过RNA深度测序,我们研究了SMA类脊髓器官(SCOs)中剪接事件的变化,并评估了吗啉寡核苷酸(MO)处理引起的剪接修饰。我们分析了对照组(CTRL,n=3个细胞系,3个独立重复样本)、SMA组(n=2个细胞系,4个独立重复样本)和经处理的SMA类脊髓器官(n=2个细胞系,4个独立重复样本),这些样本经过两个月的分化期,并使用r6-MO和r6- scramble-MO作为阴性对照进行处理(补充图14)。
在SMA与CTRL类脊髓器官(SCOs)的对比中,共发现666个基因上调、463个基因下调,证实存在整体转录改变(补充图14a及补充数据6)。基因本体(GO)分析显示,进行性脑病相关基因富集,而突触发生和谷氨酸受体信号相关基因则下调(补充图14b)。这些发现与SMA类脊髓器官所呈现的功能活性改变相关。由于 bulk组织RNA测序反映的是整个类器官的平均基因表达模式,我们利用单细胞表达数据来推断基因模块,并了解特定细胞类型中的共表达情况。该分析识别出5个不同的模块,其基因在特定细胞类型中富集:循环细胞(M1:CDK1、CDKN3、NUSAP1、TOP2A、UBE2S)、祖细胞(M2:BTG1、EFNB1、HES2/4、TUBA1C)、星形胶质细胞和祖细胞(M3:GABBR2、NTRK2、PHGDH、PTPRZ1、SLC1A4)、中间祖细胞/早期运动神经元(M4:FOXP4、RARA、SP1、TEAD2/3、TCF3)以及成熟运动神经元(M5:GRIA1/2/4、MAPT、NEFL、SCN2A)(补充图14c及补充数据6)。这些数据进一步表明,存在一种广泛的表型,涉及不同的发育动态,其中运动神经元以及正在分化的细胞均受到SMN1缺失的显著影响。
为了揭示r6-MO处理下游的分子机制,并阐明导致功能恢复的通路,我们比较了经r6-MO处理的SMA类脊髓器官(SCOs)与经r6-scramble-MO处理的SMA类脊髓器官的转录和剪接图谱。r6-scramble-MO处理作为阴性对照,为与r6-MO处理的比较提供了基线。与r6-scramble-MO处理相比,r6-MO处理后检测到的转录变化相对较少,这表明至少在所考虑的时间窗口内,表型恢复不需要全局转录变化。
相比之下,对脊髓性肌萎缩症(SMA)与对照(CTRL)、r6-MO处理与r6-MO- scramble处理的SMA类器官中前体mRNA剪接模式的分析显示,不同条件下存在显著差异(图6a、b及补充图14d)。SMA脊髓类器官(SCOs)中SMN1的缺失(对比CTRL与SMA类器官)以及r6-MO处理对SMN的挽救作用(对比r6-scramble-MO与r6-MO)导致了多种显著的可变剪接变化。变化最显著的事件为外显子跳跃(SE)、互斥外显子(MXE),其次是内含子保留(IR)、可变3’剪接位点(A3SS)和可变5’剪接位点(A5SS)事件,这些事件的检测频率较低(补充数据7)。 为评估r6-MO处理对SMA细胞剪接图谱的影响,我们将r6-MO处理的SCOs与r6-scramble-MO(基线对照)处理的细胞中的前体mRNA剪接事件进行了比较。r6-MO特异性修饰了共计2614个剪接事件(图6c及补充数据8)。其中数量最多的是SE、MXE和IR,其比例与CTRL和SMA对比中观察到的比例相似。我们鉴定出1982个与这2614个挽救性剪接事件特异性相关的基因(补充数据8)。对这些基因的基因本体富集分析显示,它们在染色质组织调控、DNA代谢过程、细胞周期以及轴突导向和神经退行性变等方面显著富集(图6c)。 重要的是,剪接事件的变化可与单细胞RNA测序分析中的特定基因模块相匹配(图6d)。我们利用所有检测到的挽救性剪接基因进行模块评分分析,揭示了与以下细胞类型特异性相关的剪接基因的独特组合:循环细胞(M1:BRIP1、CENPK、HMMR、TOP2A、UBE2C)、祖细胞(M2:ALG3、EIF5、LTN1、MRPL21、MRPL13)、祖细胞和神经元(M3:AHI1、CEP290、KIAA0586、VPS13C、XPO1)、星形胶质细胞(M4:BMPR1B、DDX3X、PDGFRB、SIRT2、SLIT2)以及运动神经元(MN)细胞类型(M5:GRIA2、MAP2、SCN1A、SCN3B、STMN4)(图6d)。
我们的研究结果表明,r6-MO处理后出现了谱系特异性的剪接改变(补充数据9)。为了评估这些变化是否能将特定基因的剪接情况恢复至对照水平,我们比较了与脊髓性肌萎缩症(SMA)表型相关的剪接事件(与对照组相比)和由r6-MO诱导的剪接事件(使用r6- scramble-MO作为特异性处理对照)。为此,我们对SMA与对照组以及r6-MO与r6- scramble-MO条件下诱导的剪接事件进行了交集分析。这种综合分析揭示了r6-MO处理后被挽救的一组独特剪接事件(图6e和补充数据9)。值得注意的是,这一类别之外的基因(在r6-MO处理后其剪接模式未完全恢复)与更广泛的病理生理过程相关,这进一步表明其系统参与度超出了脊髓疾病的范畴(补充数据10)。早期r6-MO处理恢复了98个基因的剪接异构体(补充数据9),这些基因在我们的生物信息学分析中被确定为主要候选基因。这些基因可能在减轻SMA的表型缺陷方面发挥关键作用,其中包括ARIH2、ATG4A/B、CHD9和HDAC6在内的几种染色质重塑酶成为关键参与者。随着时间的推移,这些靶点可能有助于重建祖细胞和神经元的分子特征,从而促进观察到的细胞和功能恢复。
在获救基因中,我们重点关注了CHD9(染色质域解旋酶DNA结合蛋白9)——它是胚胎干细胞中细胞周期的关键调节因子³⁸,以及HDAC6(组蛋白去乙酰化酶6)——其通过在轴突运输、自噬促进和神经保护中的作用,对运动神经元的发育和功能至关重要³⁹,⁴⁰(图6f,剪接示意图)。这些发现表明,获救的剪接事件会影响对祖细胞和神经元发育至关重要的多种细胞和分子过程,从而推动r6-MO处理后观察到的功能恢复。
特定的救援事件
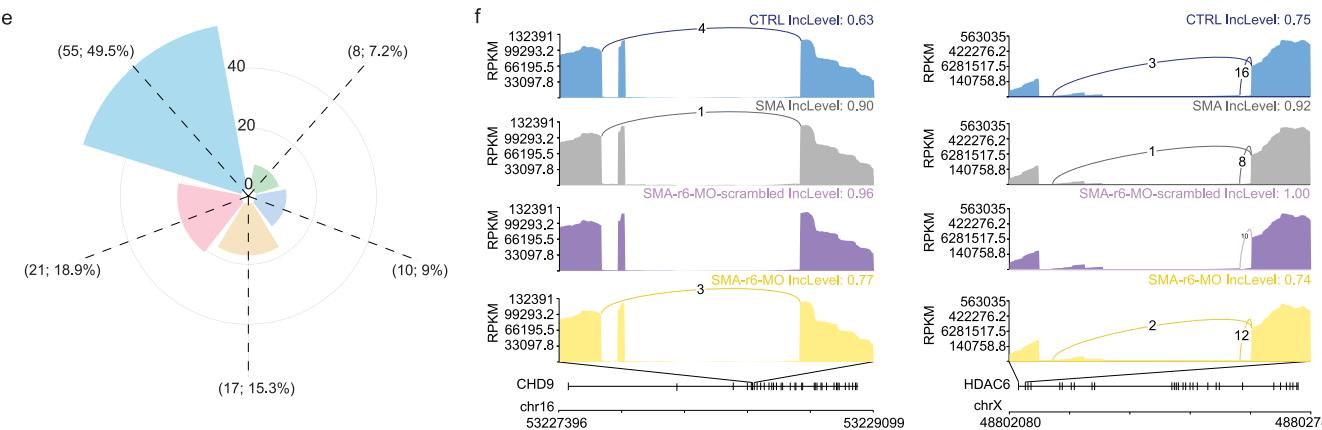
图6 | r6-MO处理可调节SMA类脊髓器官(SCOs)中的剪接事件。a 可变剪接事件(AS)主要类别的示意图。b 基于SMA与对照组(CTRL)的比较(n=3个C2,n=3个C3;n=3个S1,n=3个S2,n=3个S3),按主要事件类别分类的AS事件频率玫瑰图。c 基于r6-MO与乱序MO在SCOs中处理的比较(n=3个S1,n=3个S2,n=3个S3),按主要事件类别分类的AS事件频率玫瑰图。对与r6-MO特异性剪接事件相关的1982个基因进行基因本体论分析(使用Metascape进行)。d 聚类热图显示在SMA单细胞RNA测序中,2个月时907个具有r6-MO特异性剪接事件的基因的单细胞共表达谱,这些基因被组织成不同的模块。基因根据低维空间中共享的共表达谱被分组到模块中。单个模块的UMAP上突出显示了模块评分,每个模块都突出显示了关键基因。e 基于SMA与对照组以及r6-MO与乱序MO比较之间的重叠,按主要事件类别分类的AS事件频率玫瑰图。这些AS事件代表了在SCOs中被r6-MO处理特异性逆转的事件。f 刺身图显示了四个实验组中CHD9和HDAC6中代表性外显子跳跃事件的读数分布和剪接连接。
值得注意的是,尽管这些“逆转”基因在我们的单细胞数据集中没有表现出任何类别特异性表达,但它们在“SMA-S1不匹配簇”中显著富集,这些簇包含仅存在于一名SMA患者中的祖细胞和神经元(补充图14e、f)。尽管仅在出现这些不匹配群体的其中一个细胞系中得到证实,但这些数据进一步表明,由SMN1缺陷引起的早期剪接缺陷可能会影响脊髓发育的分子动态,破坏神经元成熟和功能的生理轨迹。
总体而言,这些结果表明,在脊髓性肌萎缩症(SMA)细胞中,r6-MO处理后发生的关键分子事件之一是恢复与脊髓发育相关基因的正常剪接模式。这也意味着,在SMA类器官中,异常的剪接动态会损害祖细胞和神经元的发育及功能,而r6-MO诱导的剪接校正因此会驱动和/或有助于挽救它们的细胞表型和功能表型。
¶ 讨论
基于干细胞的人类中枢神经系统模型为研究其他实验方法无法探究的人类生物学过程提供了机会41–43。脑类器官已被提议作为研究人类大脑发育和识别疾病表型的平台42,44–46。然而,文献中对脊髓样类器官的描述较少16,47–49,在运动神经元疾病以及存在迫切未满足治疗需求的神经系统疾病背景下,这一研究领域值得进一步探索。在此,我们利用类器官技术,从健康受试者和1型脊髓性肌萎缩症患者的诱导多能干细胞中生成脊髓类器官和脑类器官,以加深对脊髓性肌萎缩症的理解并测试创新的优化疗法。
脊髓性肌萎缩症(SMA)疾病修饰疗法的出现改变了临床实践的格局,为患者、护理人员和医疗服务提供者带来了新的希望。现有证据表明,早期治疗对临床疗效至关重要。因此,加深对该疾病产前和症状前阶段的了解,对于优化治疗策略而言极为关键。尽管SMA是一种常染色体隐性遗传病,且常在新生儿期发病,但针对该疾病产前阶段的研究却寥寥无几——而在这一阶段,SMN蛋白的功能至关重要[8]。迄今为止,尚无关于人类SMA脊髓在单细胞分辨率下的分子数据报道,也未探究过不同细胞群受到的影响。产前运动神经元的死亡和功能障碍可能会影响产后疾病的发作和严重程度,并决定治疗窗口。SMN蛋白缺乏是否会损害早期神经元发育,进而引发神经元死亡,这一问题在很大程度上仍未得到探索。从这一角度来看,SMA类器官是着手研究关键早期病理特征之间联系的理想模型。在此,我们证实,对照组(CTRL)和SMA脊髓类器官(SCOs)均包含一系列脊髓神经元结构域,重现了人类发育中脊髓内神经元群体的体内复杂性(图1)。此外,我们发现,SMA脊髓类器官的早期分化程序在细胞死亡变得显著之前就已受到影响(图2)。我们观察到,SMA脊髓类器官中处于早期发育状态的细胞数量更多(图2)。尽管基于单个时间点的拟时序推断存在局限性,但该分析仍为与SMA发病机制相关的异常发育轨迹提供了有意义的见解。这些数据,连同在细胞死亡增加的同一阶段(2个月,图3)在祖细胞和神经元类型中观察到的普遍分子改变,表明SMA发病机制中存在重要的发育因素,并提示异常的发育轨迹可能会对不同神经元亚型的分化和存活产生负面影响。
尽管与脊髓性肌萎缩症(SMA)相关的发育表型始终可检测到且具有统计学显著性,即使在对单个基因型进行分析时也是如此,但脊髓类器官(SCO)系统的一个局限性在于不同基因型间不同细胞群的代表性存在差异,而且需要承认的是,诱导多能干细胞(iPSC)系之间的运动神经元(MN)丰度存在一些差异,尤其是在对照系中。我们认识到,对直接来源于患者的诱导多能干细胞系进行分析具有使用患者特异性材料的优势,从而涵盖更广泛的生物学变异性。因此,重要的研究发现更有可能具有临床相关性。另一方面,使用同基因系可以更精确地将表型差异归因于疾病背景,而非个体间差异,因此,这为我们的研究增加了一个潜在的局限性。尽管如此,尽管存在这些限制,发育改变仍是一种普遍存在且具有生物学意义的表型。最近的一项平行研究使用诱导多能干细胞系及其相应的同基因对照构建了SMA模型,生成了包含中胚层和神经谱系的脊髓类器官49。纵向单细胞转录组分析显示神经干细胞的特化受损,且向中胚层和肌肉祖细胞偏移,这代表了在SMA早期小鼠胚胎中也观察到的发育偏向。这些发现补充了我们的研究结果,并与“SMA的发病机制源于发育过程,不仅涉及早期命运限制决定,还涉及可能导致后期运动神经元退化的更广泛的神经发育缺陷”这一假设相一致。最近对亨廷顿病等神经退行性疾病的发育因素进行了研究50-52,这表明该假设也可在SMA中进行研究。
与对照组相比,SMA类脊髓器官中神经突长度缩短的观察结果表明,该3D模型能够重现SMA的关键疾病特征(图3)。有趣的是,SMA类脊髓器官中重现了早期神经元分化缺陷和细胞凋亡,这表明分化缺陷与退化加剧同时存在可能参与了SMA的发病机制。
有趣的是,当我们研究差异表达基因(DEGs)时,发现其中很大一部分不仅在运动神经元(MNs)中被检测到,还存在于其他细胞亚群中。这些数据表明,尽管脊髓运动神经元功能缺陷是脊髓性肌萎缩症(SMA)病理的核心特征,但该疾病的影响更为广泛。越来越多的证据表明,SMA是一种多因素疾病,不同细胞类型与疾病机制之间的相互作用会逐渐导致运动神经元功能障碍和死亡⁵³,⁵⁴。磁共振成像显示,存活超过1岁的重度SMA患者会出现进行性、弥漫性脑损伤¹⁰。动物研究中已报道了产前脊髓的改变⁵⁴,最近在人类中也有相关发现⁹。一项最新研究表明,有症状的SMA小鼠的运动皮层和小脑 存在功能改变⁵⁵。在本研究中,我们发现SMA脑类器官的神经元活动受损(图4)。这种更广泛的影响需要进一步明确特征,并且在SMA患者的有效临床管理中需要加以考虑。实际上,脊髓类器官(SCOs)和脑类器官的自发活动和诱发活动的产生均受到损害(图4),这表明该模型具有生物学相关性,同时也说明发育异常和/或可塑性事件可能对神经元连接产生直接影响。在SMA³⁰,⁵⁶和肌萎缩侧索硬化症(ALS)⁵⁷中均已发现神经元过度兴奋,这与上下运动神经元的变性有关⁵⁸,⁵⁹。然而,目前尚不清楚这种功能障碍在SMA病程中出现的时间有多早,以及它在中枢神经系统各部分之间的差异有多大。SMA脊髓类器官早期就表现出基础活动降低和过度兴奋,这可能与疾病表型有关。此外,我们发现多个神经元群体中谷氨酸通道基因的调控异常依赖于SMN蛋白,这可能表明SMA神经元中与观察到的过度兴奋相关的内在转录变化。
过去十年中,脊髓性肌萎缩症(SMA)的可用疗法数量不断增加。然而,经FDA/EMA批准的治疗药物诺西那生钠(nusinersen)是一种反义寡核苷酸(ASO),它通过纠正SMN2的剪接来增加SMN的表达,但其给药需要通过定期腰椎穿刺进行。细胞穿透肽(CPPs)是一类小型阳离子分子,具有大分子化合物的载体能力,可促进反义寡核苷酸(ASO)递送时的血脑屏障通透性和组织分布。这种策略在多种疾病模型中已显示出更好的全身分布效果,包括静脉注射给药的情况,并已在杜氏肌营养不良症的研究中被提出。在本研究中,我们在SMA类脊髓器官(SCOs)的人体组织中测试了相同的偶联物,结果表明,在提高SMN水平和改善SMA病理特征方面,该药物的效果优于未偶联的吗啉寡核苷酸(MO)(图5)。
我们利用深度测序批量RNA测序技术,对脊髓性肌萎缩症(SMA)类器官中发生改变的剪接事件进行了全局分析(图6)。这使我们能够评估在不同遗传背景下剪接变化的存在情况,以及SMN校正如何在不同细胞类型层面上产生广泛影响,且这种影响与患者特异性背景无关。事实上,以单细胞RNA测序数据为参考,我们能够评估这些被挽救的靶标在处于不同发育状态的不同细胞群中是如何表达的,并揭示与SMA发病机制中脊髓发育相关的重要分子事件。在此,我们鉴定出了与SMA表型相关的剪接改变,对这些改变的校正可能有助于挽救关键的病理功能障碍。
这不仅为全面剖析在脊髓性肌萎缩症(SMA)多方面特征中治疗作用的机制开辟了新的研究途径,还证实了这些模型是改进当前临床治疗策略的合适工具,甚至有望在未来扩展到更具个性化的医疗领域。总之,我们的研究表明,SMN是产前脊髓和大脑发育过程中的关键分子,这意味着如果在该疾病的症状前阶段(即围产期甚至产前)进行干预,脊髓性肌萎缩症的治疗将会最为有效。进一步研究SMN缺乏在发育过程中对多系统的影响,对于确定最佳治疗窗口以及评估疗法对早期发育缺陷的疗效至关重要。
¶ 方法
涉及人类样本的研究是按照《赫尔辛基宣言》的伦理标准、国家法规和机构指南进行的。人成纤维细胞系来自欧洲生物样本库,是在获得知情同意的情况下获取的,并经IRCCS卡格兰达基金会大医院和米兰大学伦理委员会批准(编号0004520)。
¶ 诱导多能干细胞重编程与培养
对照诱导多能干细胞系C1和C2来源于皮肤活检获取的成纤维细胞,这些成纤维细胞使用CytoTune iPS 2.0试剂盒(CytoTune-iPS仙台2.0重编程试剂盒,Invitrogen)被重编程为诱导多能干细胞。对照系C3来自西达赛奈医学中心的CS5NXHiCTR。对照系C4来自WiCell的iPS DF 19-9-11 TH,且先前在Corti等人的研究(文献62、63)中使用过。对照系C5先前由Corti实验室构建,并在Taiana等人的研究(文献64)中发表。脊髓性肌萎缩症系S1来自西达赛奈医学中心的CS77iSMA。脊髓性肌萎缩症系S2来自西达赛奈医学中心的CS32iSMA。脊髓性肌萎缩症系S3来自西达赛奈医学中心的CS83iSMA。脊髓性肌萎缩症系S4先前由Corti实验室构建(文献65、66)。脊髓性肌萎缩症系S5先前由Corti实验室构建(文献62)。所有诱导多能干细胞系均来自男性供体。诱导多能干细胞在E8培养基(Essential 8™培养基,Gibco,A1517001)中、37°C、5% CO₂的条件下培养。
¶ 类胚体(EBs)的形成
类胚体(EBs)由在E8培养基(Essential 8™培养基[Gibco,A1517001])中培养的诱导多能干细胞(iPSCs)生成,培养条件为37°C、5% CO₂。当细胞融合度达到80–85%时,使用2 mg/mL的IV型胶原酶(Gibco,17104019)分离诱导多能干细胞集落,收集后轻轻重悬于完全类胚体培养基中。该培养基成分为:DMEM/F12(Gibco,11320033)、KnockOut血清替代物(Gibco,10828028)、MEM非必需氨基酸溶液(Gibco,11140035)、55 mM的2-巯基乙醇(Gibco,21985023)、GlutaMAX(Life Technologies,35050061),并添加4 ng/mL的碱性成纤维细胞生长因子(Peprotech,100-18B)。将细胞转移至未处理的组织培养 Petri 皿中以促进悬浮生长。类胚体在悬浮状态下培养长达7天,根据制造商说明(编号MAN0013784 修订版1.0),每隔一天更换一次培养基(不含碱性成纤维细胞生长因子的类胚体培养基)。
¶ 大脑类器官(CrOs)的生成
脑类器官的生成采用了先前的改良方案¹⁴。使用Accutase(Life Technologies公司,货号A1110501)收集选定的诱导多能干细胞(iPSCs),将约9000个iPSCs接种到超低吸附96孔板(Corning公司,货号CLS7007)的每个孔中,培养基为多能干细胞培养基,其中含有低浓度的重组人碱性成纤维细胞生长因子(4 ng/mL,Peprotech公司,货号100-18B)和Y-27632(50 μM,ROCK抑制剂,Calbiochem公司,货号13624S)。第6天,将获得的类胚体(EBs)转移到低吸附24孔板(Corning公司,货号CLS3473)中,使用神经诱导培养基(NIM)培养,该培养基包含DMEM/F12(Gibco公司,货号11320033)、1% N2补充剂(Gibco公司,货号17502001)、1% GlutaMAX(Life Technologies公司,货号35050061)、1% MEM非必需氨基酸(Gibco公司,货号11140035)和1 μg/mL肝素(Sigma公司,货号H3149)。第12天,将类胚体转移到Cultrex® 2型基底膜基质(BME type 2,Trevigen公司,货号3532-001-02)的液滴中,在分化培养基中生长;分化培养基由DMEM/F12和Neurobasal(Gibco公司,货号21103049)按1:1混合而成,含有0.5% N2补充剂、1%不含维生素A的B27补充剂(Gibco公司,货号12587010)、2-巯基乙醇、0.025%胰岛素(Sigma公司,货号I9278)、1% GlutaMAX和0.5% MEM非必需氨基酸。2天后,将液滴转移到旋转生物反应器中,所用培养基与上述相同,但不含维生素A的B27补充剂替换为含维生素A的B27补充剂(Gibco公司,货号17504044)。旋转生物反应器的转速逐渐提高至45 rpm。
¶ 脊髓类器官(SCOs)的生成
为了生成SCOs,从第0天到第2天采用了与人类大脑类器官相同的操作流程。第2天,将一半培养基更换为新鲜培养基,其中包含终浓度为4 ng/mL的bFGF(Peprotech公司,货号100-18B)、50 μM的Y-27632(ROCK抑制剂,Calbiochem公司,货号13624S)、3 μM的CHIR 99021(GSK3抑制剂,Sigma-Aldrich公司,货号SML1046)、2 μM的SB-431542(激活素/BMP/TGF-β通路抑制剂,Sigma-Aldrich公司,货号S4317)以及0.2 μM的LDN-193189(骨形态发生蛋白[BMP]通路抑制剂,Stemcell Technologies公司,货号72149)。第4天,更换培养基,并添加相同的小分子物质(不含bFGF和Y-27632)。第6天,将EBs转移至超低吸附24孔板中,在NIM培养基中添加100 nM视黄酸(RA,Sigma-Aldrich公司,货号R3255)进行培养。第12天,将EBs包埋在Cultrex中,使用SCO分化培养基(SCODM,由1:1的DMEM/F12和Neurobasal培养基组成,添加0.5% N2补充剂、1% B27、2-巯基乙醇、0.025%胰岛素(Sigma公司,货号I9278)、1% GlutaMAX和0.5% MEM-NEAA)进行培养。向SCODM中加入1 μM的Smoothened激动剂(SAG,Calbiochem公司,货号364590-63-6)。第14天,将SCOs转移至旋转生物反应器中,使用添加了RA和SAG的SCODM进行培养。第28天,将RA和SAG的浓度降至50%,并添加神经营养因子BDNF(Peprotech公司,货号450-02)、GDNF(Peprotech公司,货号450-10)和IGF-1(Sigma-Aldrich公司,货号100-12),每种因子的浓度均为10 ng/mL。
¶ 免疫组织化学分析
类器官在室温(RT)下用4%多聚甲醛(PFA)固定15分钟,随后置于含30%蔗糖的1×PBS中,在4°C下过夜。次日,将其包埋于7.5–10%明胶/蔗糖中,用干冰冷冻,在−80°C下过夜储存,然后使用冷冻切片机将其切成20μm的切片。类器官切片在室温下用含0.3% Triton X-100(Sigma)和10%驴血清或山羊血清(Jackson ImmunoResearch)的1×PBS透化1小时。二维培养物在室温下用4% PFA固定5分钟,然后在室温下用含0.2% Triton X-100(Sigma)和10%驴血清或山羊血清(Jackson ImmunoResearch)的1×PBS透化1小时。将稀释在新鲜透化溶液中的一抗在4°C下孵育过夜(完整列表汇总于补充数据2)。次日,在室温下,将二抗(AlexaFluor 488、555、568、594和647偶联物,1:1000;Invitrogen)与DAPI(1:500;Sigma-Aldrich,D9542)一起加入1×PBS中,孵育90分钟。使用DAKO封片液(Agilent,S3023)封盖盖玻片。TUNEL测定使用DeadEnd™荧光TUNEL系统(Promega,G3250)进行。共聚焦图像使用Leica SP8白光激光倒置共聚焦显微镜(Leica)获取,并使用Fiji图像处理软件进行分析。
¶ 图像量化
为了评估SOX2⁺细胞的数量和TUJ1阳性区域的百分比,使用Leica DMi8倒置显微镜和Leica LAS X软件平台获取了整个SCO切片的10×拼接图像。使用Zeiss Axio倒置显微镜和ZEN Light软件平台获取了整个脑类器官切片的20×拼接图像。每个类器官至少获取3个不同的切片,每个细胞系至少使用3个类器官,因此CTRL组和SMA组的样本量均大于3(补充数据1)。具体而言,为了计算SOX2⁺细胞的数量,设计了一个定制的Cell Profiler⁶⁷流程,并应用定制设计的ImageJ/Fiji⁶⁸图像宏来评估TUJ1⁺区域的比例(%)。对于TUNEL定量,使用尼康Crest倒置多模态转盘共聚焦显微镜获取图像。使用NIS-Elements v5.21软件(Nikon-Lim Instruments)中的GA2模块,通过专门设计的阈值分割分析流程对细胞核(DAPI)和TUNEL阳性细胞进行定量。关于神经丝追踪,使用Fiji图像处理软件对之前用Leica SP8白色激光倒置共聚焦显微镜(Leica)获取的图像进行分析。使用NeuronJ插件对神经突长度进行追踪和分析;仅追踪那些起点和终点均在视野内的丝状体。延伸到视野外或无法确定起点的丝状体不纳入分析。
¶ 实时定量聚合酶链式反应(qRT-PCR)
使用ReliaPrep RNA细胞小量提取系统(Promega,Z6010)提取总RNA。使用Nanodrop(赛默飞世尔科技)检测RNA浓度。使用SuperScript IV VILO Master Mix(赛默飞世尔科技,11756500)进行逆转录,将150 ng模板总RNA逆转录。该方案已用于处理大量RNA样本。候选基因(补充数据3)的表达水平通过以下方法评估:(i)在7500实时PCR系统和7900HT快速实时PCR系统(Applied Biosystem)上进行TaqMan定量分析,以GAPDH作为参考基因;(ii)在ViiA 7实时PCR系统(赛默飞世尔科技)上进行Powertrack SYBR(赛默飞世尔科技)定量分析,以ACTB作为参考基因。
¶ FL和Δ7转录本分析
使用SuperScript IV VILO Master Mix(赛默飞世尔科技)对从诱导多能干细胞(iPSCs)或脊髓小脑共济失调类器官(SCOs)中提取的总RNA进行逆转录。通过对逆转录聚合酶链反应(RT-PCR)产物的限制性片段长度多态性(RFLP)分析来评估FL和Δ7转录本的相对表达水平(引物可按需索取)。聚合酶链反应(PCR)产物经DdeI内切核酸酶在37°C下过夜消化,随后在2.5%琼脂糖凝胶上进行电泳。DdeI可识别并切割含有外显子8的分子,从而能够区分FL和Δ7亚型的SMN1和SMN2基因组来源。
¶ 电生理学
我们使用高密度微电极阵列(HD-MEA)记录脊髓类器官(SCOs)和大脑类器官的细胞外自发活动。该设备包含4096个平面电极,每个电极尺寸为21×21 μm,覆盖面积为2.67×2.67 mm(采用瑞士3Brain AG公司的Biocam X系统及Biochip Arena芯片)。将类器官轻轻放置在芯片上,并用铂锚和尼龙网固定。记录前后及记录过程中,类器官均浸泡在克雷布斯溶液中,该溶液成分如下(mM):120 mM氯化钠、2 mM氯化钾、1.19 mM硫酸镁、26 mM碳酸氢钠、1.18 mM磷酸二氢钾、11 mM葡萄糖、2 mM氯化钙,用95%氧气和5%二氧化碳的混合气体充氧,使pH值达到7.4。 首先在对照条件下记录类器官15分钟,然后加入100 μM L-谷氨酸(谷氨酸,Sigma-Aldrich公司)后再记录15分钟,之后加入100 μM谷氨酸、10 μM Gabazine(SR-95531,Abcam公司)和1 μM盐酸士的宁(Sigma-Aldrich公司)的混合溶液,继续记录15分钟。使用Brainwave X软件(3Brain AG公司)以17 kHz的采样频率记录电生理活动,采用Brainwave 4软件(3Brain AG公司)进行离线轨迹分析。 采用精确时间峰值检测算法检测峰值(所用参数:正负峰值、峰值持续时间<1 ms、不应期>1 ms、检测阈值>噪声标准差的7倍)。通过主成分分析结合K均值聚类(轮廓系数法)进行峰值分类,每根电极最多考虑3个聚类,每个聚类最多3个峰值。使用Pakhira-Bandyopadhyay-Maulik指数验证聚类结果⁶⁹。研究人员通过重新分析信号并去除所有干扰因素进行最终检查。仅考虑在对照期间表现出稳定活动的通道(峰值频率波动不超过20%)。 用于分析的每个类器官的通道数量在对照类器官(脊髓类器官:13.6±0.6;大脑类器官:13.8±2.1)、脊髓性肌萎缩症(SMA)类器官(脊髓类器官:15.2±0.7;大脑类器官:15.4±0.9)和经治疗的脊髓性肌萎缩症类器官(脊髓类器官:12.8±1.3)之间具有可比性。基础放电频率的百分比变化通过比较加入谷氨酸前最后10分钟的平均放电频率与加入谷氨酸后5至15分钟期间的平均放电频率计算得出。
¶ 吗啉代处理
我们使用了一种吗啉代(MO-10–34)序列,该序列靶向SMN2外显子7下游的ISS-N1区域,这是我们实验室之前描述过的³⁴。设计用于靶向SMN2前体mRNA的特定MO序列为GTAAGATTCACTTTCATAATGCTGG。本研究中使用的细胞穿透肽(CPP)是r6(RRRRRR,D对映体构型),由我们的外部合作者Hong Moulton博士将其共价偶联到MO的5’端。 scrambled MO(scr,GTAACATTGACTTTGATATTCCTGG)被用作反义特异性的对照。我们使用了单独的MO以及与r6肽偶联的MO(r6-MO)³⁶。作为阴性对照,我们使用了一种含有scrambled序列的定制r6-MO。为了评估生物分布,我们使用了生物素化的r6-MO,并按如下方法将其添加到培养基中。所有的MO寡聚物均由Gene Tools合成。我们用浓度递增的MO或r6-MO处理SMA类脊髓器官(SCOs)。将含有2.4纳摩尔MO或r6-MO的溶液直接加入每个孔中,孵育48小时。之后,更换培养基,并再次加入2.4纳摩尔的MO或r6-MO,继续孵育48小时。然后,将培养基更换为新鲜培养基,持续72小时。最后,再次用4.8纳摩尔的MO或r6-MO处理类器官48小时。
¶ 蛋白质印迹分析
蛋白质印迹分析按照先前描述的方法进行³⁴。简要来说,样本在添加了蛋白酶和磷酸酶抑制剂(Pierce,美国伊利诺伊州罗克福德)的裂解缓冲液中于冰上超声处理10分钟。约25μg蛋白质通过12% SDS-PAGE分离,并电泳转移至硝酸纤维素膜上。膜与抗SMN抗体(1:1000,BD)和抗β-肌动蛋白抗体(1:1000,Sigma,美国密苏里州圣路易斯)孵育。用辣根过氧化物酶偶联的二抗(Life Technologies)标记后,使用化学发光底物(Amersham,美国宾夕法尼亚州匹兹堡)检测蛋白质。信号通过LI-COR Odyssey 9120成像系统获取。使用Image Studio Lite(v5.2)进行光密度分析。
¶ 来自解离类器官的细胞的二维培养
将小肠类器官(SCOs)以300×g离心5分钟,加入胰蛋白酶-EDTA后,将类器官转移至37°C水浴中放置5分钟。随后对类器官进行机械解离,加入两倍体积的马血清(Euroclone公司,货号ECS0091L)以灭活胰蛋白酶,之后进行离心。弃去上清液,用小肠类器官分化培养基(SCODM)重悬沉淀。将约50,000个细胞接种到24孔板中,孔内含有预先用多聚-L-鸟氨酸(Sigma-Aldrich公司,货号P3655)和层粘连蛋白(Sigma-Aldrich公司,货号L2020)包被的盖玻片。使用添加了视黄酸(RA)、环巴胺类似物(SAG)和生长因子的小肠类器官分化培养基(SCODM)来维持类器官来源的细胞,其中添加物的浓度与分化第28天时使用的浓度相同。
¶ 全标本染色
类器官用1×PBS洗涤,然后在4%多聚甲醛(PFA)中于4°C的旋转轮上固定过夜。次日,在室温(RT)下的旋转轮上用1×PBS洗涤五次(每次10分钟)。类器官用含0.25% Triton X-100的1×PBS透化15分钟。为防止非特异性结合,将类器官在含10%胎牛血清(FBS)和0.5%与一抗同物种血清的1×PBS中孵育。一抗分三轮(2小时、5小时和12小时后)依次加入,并在含5%胎牛血清、0.5%与一抗同物种血清及0.1% Triton X-100的1×PBS中稀释。二抗在与一抗相同的溶液中稀释,并在室温下于旋转轮上孵育过夜。图像由Alembic实验设施(圣拉斐尔研究所IRCCS医院)使用尼康A1R MP+多光子显微镜采集。
¶ 用于单细胞RNA测序分析的SCO细胞解离
类器官的解离采用基于Worthington木瓜蛋白酶解离系统试剂盒(Worthington Biochemical)的优化方案进行。单细胞悬液收集在含0.04% BSA(Sigma-Aldrich)的冰冷1×PBS(无钙镁)中,通过自动细胞计数仪(Countess II,赛默飞世尔)计数,密度为1×10⁶细胞/毫升。
¶ 单细胞RNA测序及下游分析
样本处理。使用单细胞3’ v3试剂套装(10× Genomics),将每个样本中约8000个细胞(按上述方法制备)加载到单细胞芯片B的一个通道中,在Chromium系统中生成凝胶珠乳液。捕获和裂解后,按照制造商的协议(10× Genomics)进行cDNA合成并扩增14个循环。然后,每个样本使用扩增后的cDNA(50 ng)构建Illumina测序文库。按照10× Genomics的读数生成说明,在Illumina NextSeq 550测序平台上进行测序。每个样本的测序深度约为45,000条读数/细胞。
数据分析。 使用整合到CellRanger(10× Genomics)套件(版本=3.0.2)中的Illumina bcl2fastq工具,将原始测序数据(bcl文件)转换为fastq文件。从原始数据开始,使用CellRanger分析流程生成数字基因表达矩阵。将读数比对到人类GRCh38参考基因组,进行注释,并使用Ensembl版本93的基因注释进行计数。使用CellRanger计数模块,采用默认设置对读数进行映射,序列长度设置为r1-length=28和r2-length=55。每个细胞至少产生90,000个读数。为了进行进一步的预处理和分析,我们使用了Scanpy⁷⁰单细胞分析工具包(版本=1.9.6,pandas版本=2.2.0,numpy版本=1.26.3)。使用Scrublet⁷¹(版本=0.2.3)识别并去除双细胞。双细胞分数的阈值由Scrublet算法自动设置,并针对每个样本单独应用。使用sc.pp.calculate_qc_metrics函数计算质量控制指标,包括线粒体和核糖体含量,然后对数据对象进行过滤,去除含有>10%线粒体转录本和>65%核糖体转录本的异常细胞。此外,使用“n_genes_by_counts”的样本特异性中位数绝对偏差(MAD)阈值对细胞进行过滤。过滤后,使用sc.AnnData.concatenate将数据连接起来,并标注样本名称和基因型。我们使用scVI-tools⁷²(版本=1.1.0)来减轻批次效应,其中每个基因型和样本都被视为一个批次。首先通过将计数复制到adata.layers[“counts”]并使用sc.pp.highly_variable_genes(Seurat V3风格)识别2000个高可变基因(考虑“Sample”批次键)来转换AnnData对象。通过设置具有分类协变量“Sample”和“genotype”以及连续协变量“pct_counts_mt”“pct_counts_ribo”和“total_counts”的AnnData,为scVI分析准备转换后的数据。使用scvi.model.SCVI初始化scVI模型,并采用默认参数进行训练。使用model.get_latent_representation提取潜在表征,并存储在adata.obsm[“X_scvi”]中。通过使用sc.pp.neighbors构建K近邻(KNN)图(使用潜在表征X_scvi)并以min_dist=0.2运行UMAP来进行降维。使用 Leiden算法⁷³(sc.tl.leiden)在分辨率0.25、0.3、0.5、0.7和1.0下执行聚类,并通过UMAP图(sc.pl.umap)可视化结果,显示聚类分配、样本信息和基因型。整合后,使用adata.raw.to_adata将数据还原为所有基因。根据最近的单细胞RNA测序数据转换基准研究⁷⁴的建议,使用sc.pp.normalize_total(adata,target_sum=None,inplace=False)对原始计数进行归一化,并使用sc.pp.log1p进行对数转换。保存最终处理的数据用于下游分析。使用Scanpy的sc.tl.score_genes函数计算基因集分数。为了识别差异表达基因(DEGs),我们对所有已识别聚类(分辨率τ=0.7)之间的归一化计数应用Wilcoxon秩和检验;按p值和对数倍数变化排序的前50个基因被用于生物学解释。
差异丰度检验。 我们使用Milo²⁵的Python版本(v=0.1.1)进行了差异丰度检验。使用milo.make_nhoods()以0.1的比例构建邻域。使用milo.count_nhoods()并基于“Sample”样本列计算邻域计数。两种基因型(CTRL和SMA)均被编码为分类变量(连续编码不适用于基因型组)。使用milo.DA_nhoods()进行差异丰度检验,其设计公式基于基因型。使用milopy.utils.build_nhood_graph()构建邻域图,并使用milopy.plot_nhood_graph()进行可视化,突出显示1%的空间假发现率(FDR)。使用milopy.utils.annotate_nhoods()为邻域标注细胞类型,并将来自 Leiden 聚类的调色板映射到邻域上。
SCO基因型间的伪批量差异基因表达分析。 为了检测特定细胞身份(每种基因型至少占25%的细胞比例)的基因型间差异表达基因(DEGs),我们利用了PyDESeq2⁷⁵(v=0.4.4)和decoupleR⁷⁶的Python实现(v=1.6.0)。使用decoupler.get_pseudobulk()函数生成伪批量谱,每组至少包含10个细胞和1000个计数。对伪批量数据进行标准化、对数转换和缩放,然后进行主成分分析(PCA)。使用decoupler.get_metadata_associations()计算元数据与主成分之间的关联,并通过decoupler.plot_associations()将结果可视化。按细胞类型对数据进行子集划分,并对选定的细胞类型进行差异表达分析。按表达量筛选基因,要求最小计数为10且最小总计数为15。构建DESeq2对象时,设计因子为“基因型”,参考水平为“CTRL”。计算对数倍变化(LFCs),并提取SMA与CTRL基因型之间的对比。通过调整后的p值(<0.05)和绝对LFC(>0.5)筛选差异表达基因。随后使用自定义R脚本将结果可视化为复杂热图和火山图。利用Ingenuity通路分析(IPA;Qiagen Ingenuity Systems)对选定细胞类型的差异表达基因列表进行分析,以计算经典通路和生物学功能的富集情况。
SCO分化状态的预测和伪时间分析。 我们使用CellRank⁷⁷(v=2.0.0)实现的CytoTRACE⁷⁸对整合数据进行了发育潜力预测。通过确保有拼接和未拼接的RNA计数来对数据进行预处理,当这些层缺失时,从初级表达矩阵中建立。使用scvelo.pp.moments()计算拼接和未拼接丰度的矩。之后,应用CellRank的CytoTRACE核来计算CytoTRACE分数并构建相应的过渡矩阵。CytoTRACE伪时间由计算出的分数得出,用于定量表示细胞分化状态。伪时间分数以密度图的形式可视化,用于比较不同的基因型。使用高斯核密度估计(scipy.stats.gaussian_kde())估计密度分布,并以脊线图的形式绘制,按基因型和细胞类型进行颜色编码。使用Python中的Matplotlib库进行可视化和图形定制。为了统计比较不同基因型(CTRL与SMA)之间的伪时间分布,使用SciPy的ks_2samp()函数进行了双侧Kolmogorov-Smirnov(KS)检验。提取每种基因型的伪时间值,并计算KS统计量和相关的p值,以评估组间分布的差异。
运动神经元谱系的调控子推断和轨迹分析。 为了推断运动神经元(MN)细胞谱系中转录因子(TFs)与推定靶基因之间的共表达模块,我们使用了pySCENIC(v=0.12.1)流程²⁸。我们利用Singularity容器对每个SCO基因型数据对象进行了独立的流程运行。在调控子推断的第一步,该流程依赖于一个机器学习模型,该模型通过GRNBoost2⁷⁹算法使基因的表达模式与转录因子的表达模式相匹配。在这部分中,我们使用了命令行工具(pyscenic:0.12.1 arboreto_with_multiprocessing.py)并采用默认参数。接下来,输出的拟合结果被用于预测潜在的调控子和直接结合靶标,然后通过带有(pyscenic:0.12.1 pyscenic ctx)和“–max dropout”参数的转录因子基序富集分析进行优化。最后,分别通过AUCell方法⁸⁰以及(pyscenic:0.12.1 pyscenic aucell)对SMA和CTRL数据对象中每个调控子的活性进行细胞评分。获得的调控子活性评分和元数据被用于可视化和轨迹分析的二次分析中。
我们使用Palantir⁸¹(版本=1.3.3)和scFates⁸²(版本=1.1.1)对聚焦于运动神经元谱系的SCOs进行了轨迹分析。加载SCO数据对象后,移除了与运动神经元谱系无关的特定集群。计算扩散图以生成具有四个特征向量的多尺度扩散空间。使用scf.tl.tree()构建主树,参数为:method=“ppt”、nodes=200、ppt_lambda=100、ppt_sigma=sig、ppt_nsteps=200。使用scf.tl.root()将根设置为SOX2表达,其对应于pMN(推定运动神经元)群体。之后,使用scf.tl.pseudotime()计算伪时间,参数为:n_jobs=20、n_map=50、seed=42。为探究运动神经元在共享轨迹上分化的差异,将数据按基因型分为CTRL和SMA运动神经元组。对于每组,使用scf.pl.dendrogram()和KDE图(seaborn.kdeplot())生成树状图。接下来,处理pySCENIC AUCell输出,使其与之前生成的运动神经元轨迹数据对齐,该方法采用自Petitpré C等人的研究⁸³。从.loom文件加载对照组和SMA样本的pySCENIC AUCell输出。通过匹配细胞ID,使AUCell矩阵与轨迹数据对齐。过滤掉在少于6个细胞中活跃的调节子。使用scFates和GAM(scf.tl.test_association(),参数为n_jobs=20和A_cut=0.025)测试调节子活性与轨迹数据的关联性。基于细胞在轨迹分析中的顺序(adata.uns[“pseudotime_list”])更新细胞的伪时间,并使用scf.tl.fit()(n_jobs=20)拟合轨迹模型。使用scf.pl.single_trend()和scf.pl.trends()可视化特定调节子活性沿轨迹片段的趋势。对于对照样本,突出显示一组显著的调节子,并绘制这些调节子的趋势。对SMA样本进行了类似分析。
信息性剪接基因模块的识别。 为了将剪接基因模块与SCO对象(CTRL或SMA)中的簇相关联,我们对该对象进行子集化处理,仅保留选定的剪接基因池(所有细胞均被保留)。在进行scVI降维后,我们通过计算基因的自相关性并筛选FDR<0.05的显著基因,开展了Hotspot⁸⁴(版本1.1.1)分析以识别基因模块。使用15个并行任务计算显著基因的局部相关性,并以50个基因的最小阈值(仅核心模式)和0.05的FDR阈值生成基因模块。计算模块得分并分配给数据集。最后,使用自定义调色板在UMAP坐标上以散点图的形式可视化剪接基因的模块得分。
阶段映射以参考胎儿脊髓数据。 为了将我们的对照类器官数据集映射到时间发育参考上,我们首先使用Scanpy处理了一个胎儿脊髓单细胞图谱²³。经过标准的细胞和基因过滤后,对数据进行标准化,并使用批量感知的“seurat_v3”方法识别出2000个高可变基因(HVG)。然后,我们在这些高可变基因上训练了一个四层scVI模型,以创建潜在的参考空间。随后,我们的对照类器官查询数据通过为缺失的基因添加零表达值,与这2000个基因集对齐。使用scArches(v = 0.6.1)软件包,我们通过部分模型再训练将查询数据投影到参考图谱上。然后,使用加权k近邻(kNN)分类器(k = 100)从图谱中转移“妊娠周数”和“ trimester”标签,推断出CTRL类器官细胞的发育阶段,并通过计算细胞存在分数来量化查询状态与参考状态之间的相似性。
细胞类型与参考胎儿脊髓数据的映射。 首先,从.h5ad文件中将胎儿脊髓图谱的原始计数矩阵²⁴加载到AnnData对象中。原始计数保存在单独的层中;然后将数据标准化为每个细胞10,000个总计数(sc.pp.normalize_total())并进行对数转换(sc.pp.log1p())。使用seurat_v3方法在原始计数层上跨批次识别出前2000个高变基因,并对数据集进行子集化以仅保留这些基因。为了校正与发育阶段相关的批次效应,我们使用了scVI-tools软件包(v=1.3.0)。该模型是利用原始计数和阶段注释作为批次键构建的。scVI模型初始化时设置了两个隐藏层,并通过训练来学习潜在的细胞表征。这种潜在空间嵌入是半自动细胞注释的基础。为了分配广泛的细胞身份,使用改编自参考文献24的标记基因计算基因模块分数。这些分数能够对主要组织/细胞类型进行分类,包括脊髓祖细胞、脊髓神经元、感觉神经元/祖细胞、中胚层、造血细胞和红细胞,如参考文献24中所展示的那样。
参考模型预训练。 我们使用经过预处理和注释的胎儿脊髓数据集来训练scPoli⁸⁵模型;scPoli是一种深度生成模型,旨在整合不同条件下的数据集,同时保留细胞类型的特性。我们首先使用seurat_v3方法,在不同实验批次(以发育阶段作为批次键)中识别出2000个高度可变基因(HVGs)。scPoli模型在仅包含这些高度可变基因的数据子集上进行初始化。模型配置中,将发育阶段作为条件键,手动注释的细胞类型作为细胞类型键。模型架构设定为32维潜在空间和一个包含1024个节点的单隐藏层。数据重建采用零膨胀负二项损失函数。模型最多训练50个 epoch,其中包括40个预训练 epoch,并采用早停机制,通过监测验证原型损失来决定是否停止,容忍期为10个 epoch。训练完成后,提取学习到的潜在表征并存储到原始的AnnData对象中。这种整合后的“X_scpoli”嵌入随后用于迁移学习和下游分析,包括k近邻图构建和UMAP可视化。 为了将我们的脊髓类器官数据集与体内参考数据集进行比较,我们使用基于胎儿脊髓图谱构建的预训练scPoli模型进行计算映射。首先,加载查询类器官数据集,并分离出对照(CTRL)样本细胞用于分析。为确保与参考模型的兼容性,将查询数据的基因集与参考训练所用的2000个高度可变基因进行对齐。对于参考数据中存在而查询数据中缺失的基因,以零计数表达值进行补充。我们利用scArches⁸⁶(v = 0.6.1)工作流将查询数据投影到参考潜在空间中。映射过程采用部分再训练策略(retrain = “partial”),训练30个 epoch,在保持参考细胞嵌入固定的同时,针对查询数据对模型进行微调。映射完成后,我们在共享潜在空间中构建加权k近邻(wKNN)图,通过HNOCA-tools包⁸⁷(v = 0.1.1)将每个查询细胞与其在参考图谱中k = 100个最近邻相连。该图构成了标签转移的基础:将参考细胞类型注释分配给每个类器官细胞,得到最终标签和定量置信度分数。随后,将所得的潜在空间坐标和转移的注释添加回原始查询对象中,以便进行进一步分析。其次,我们计算了原代组织的细胞存在分数,以量化类器官样本与胎儿参考图谱之间的组成相似性。所得的潜在嵌入和注释被添加到查询对象中,用于下游可视化和分析。
¶ bulk RNA测序分析
文库构建。从9个对照类器官、6个脊髓性肌萎缩症(SMA)类器官和16个吗啉代处理的SMA类器官(8个r6-MO,8个r6-MO- scramble)中提取50 ng总RNA,使用SMARTer链特异性RNA-Seq试剂盒(TaKaRa),按照制造商的说明制备文库。样本在Illumina NextSeq 2000平台上进行了高覆盖度的双端150 bp链特异性测序。
校准。 对于每个样本组(9个对照类器官、6个SMA类器官、8个经r6-MO处理的SMA类器官以及8个经r6-MO- scramble处理的SMA类器官),原始读数通过STAR v2.7.10a⁸⁸映射到人类hg38基因组并进行定量分析。
SMN1和SMN2计数。 SMN1(SMN-C)和SMN2(SMN-T)基因具有极高的同源性(>99%)。因此,为了区分来自这两个基因的读数,我们对映射到第7外显子的读数进行了计数,其中单核苷酸的差异使SMN1和SMN2能够被明确区分。这项验证是使用IGV工具进行的。映射到基因其他区域的计数被排除在本分析之外。
差异基因表达分析。 使用edgeR包(v4.0.2)在R语言中进行差异基因表达分析,涉及estimateDisp、glmFit和glmLRT函数。过滤掉在至少6个样本中每百万次计数(cpm)<0.05的基因。
基因本体论差异表达基因。 使用Ingenuity通路分析对CTRL和SMA类器官之间的差异表达基因进行了基因本体论分析。对“经典通路”和“疾病与功能”中的术语进行了筛选。
差异剪接分析。 样本间的差异剪接分析使用rMATS-turbo v4.3.0⁸⁹进行,该工具可识别5种基本的可变剪接(AS)模式:外显子跳跃(SE)、互斥外显子(MXE)、可变5’剪接位点(A5SS)、可变3’剪接位点(A3SS)和内含子保留(RI)。显著事件的特征为FDR < 0.05且绝对包含水平差异 > 0.05。分析仅使用映射到外显子-外显子连接的读段。使用rmats2sashimiplot v2.0.4(https://github.com/Xinglab/rmats2sashimiplot)生成感兴趣事件的生鱼片图。
剪接事件的基因本体论。 比较了SMA与CTRL以及SMA r6-MO与SMA r6-MO- scramble的差异剪接事件。满足(i)相同起始位置和相同终止位置以及(ii)具有相反包含水平差异的事件被称为“共同事件”。为了注释基因功能,使用Metascape90进行了基因本体论分析。剪接和基因表达数据的下游处理是在R v4.1.0中使用自定义脚本完成的。
¶ 统计分析
连续变量以均数±标准差表示。组间比较以及组内基线与干预后或不同时间点之间的比较,分别采用非配对和配对双侧Student t检验。多组间比较采用单因素或双因素方差分析,当整体检验的零假设被拒绝时,采用Tukey HSD或Sidak事后检验。所有分析均使用GraphPad Prism v8.0软件进行,显著性水平设为0.05。
¶ 报告摘要
有关研究设计的更多信息,请参见本文所链接的《自然》系列期刊报告摘要。
¶ 数据可用性
所有数据均已在正文或补充材料中提供。本论文附带了源数据。单细胞RNA测序的读长水平数据已存放在GSE290980,而批量RNA测序的计数水平数据和元数据已存放在GSE290979。本论文附带了源数据。
¶ 参考文献
- Kolb, S. J. 等。婴儿型脊髓性肌萎缩症的自然病史。《神经病学年鉴》82, 883–891 (2017)。
- 法拉韦利(I.)、尼扎尔多(M.)、科米(G. P.)和科尔蒂(S.)。脊髓性肌萎缩症——这一古老挑战的近期治疗进展。《自然评论·神经病学》,11卷,351-359页(2015年)。
- Lorson, C. L., Hahnen, E., Androphy, E. J. 及 Wirth, B. SMN基因中的一个单核苷酸可调控剪接并导致脊髓性肌萎缩。《美国国家科学院院刊》96, 6307–6311 (1999)。
- Parente, V. 和 Corti, S. 脊髓性肌萎缩症治疗的进展。《神经疾病治疗进展》11, 1756285618754501(2018)。
- Mercuri, E., Pera, M. C., Scoto, M., Finkel, R. 及 Muntoni, F. 脊髓性肌萎缩症——治疗时代的见解与挑战。《自然综述·神经病学》16, 706–715 (2020)。
- Mercuri, E., Sumner, C. J., Muntoni, F., Darras, B. T. 及 Finkel, R. S. 脊髓性肌萎缩症。《自然综述:疾病初报》8, 1–16 (2022)。
- Soler-Botija, C., Ferrer, I., Gich, I., Baiget, M. 及 Tizzano, E. F. Ⅰ型脊髓性肌萎缩症脊髓中的神经元死亡加剧且始于胎儿发育阶段。《大脑》125, 1624–1634 (2002)。
- 拉莫斯(D. M.)等人。疾病相关组织中依赖年龄的SMN表达及其对脊髓性肌萎缩症治疗的意义。《临床研究杂志》,129卷,4817–4831页(2019年)。
- Kong, L. 等。产前运动轴突发育受损需要对重度脊髓性肌萎缩症进行早期治疗干预。《科学转化医学》13, eabb6871 (2021)。
- Mendonça, R. H. 等。0型5q脊髓性肌萎缩症的严重脑受累。《神经病学年鉴》86, 458–462 (2019)。
- 法拉韦利(I.)、科斯塔马尼亚(G.)、塔马尼尼(S.)和科尔蒂(S.)。回到起源:利用人脑类器官研究神经退行性变。《脑研究》,1727期,146561(2020)。
- 蒂齐亚诺,F. D. 和蒂扎诺,E. F. SMN基因的25年:脊髓性肌萎缩症的哥白尼式革命。《肌动蛋白》39,336-344(2020)。
- Tizzano, E. F. 等人。RESTORE注册中心中患有脊髓性肌萎缩症且携带4个或更多SMN2基因拷贝并接受onasemnogene abeparvovec治疗的患者的结局。《欧洲儿科神经病学杂志》,53卷,18-24页(2024年)。
- 兰开斯特(M. A.)等人。类脑器官模拟人类大脑发育和小头畸形。《自然》501,373–379(2013)。
- Velasco, S. 等人。单个脑类器官可重复形成人类大脑皮层的细胞多样性。《自然》570, 523–527 (2019)。
- Andersen, J. 等人。功能性人类3D皮质-运动组装体的生成。《细胞》183, 1913–1929.e26 (2020)。
- 马晓然等人。TDP-43抑制额颞叶痴呆-肌萎缩侧索硬化症基因UNC13A中隐蔽外显子的包含。《自然》603,124-130(2022)。
- Sagner, A. 与 Briscoe, J. 建立脊髓中的神经元多样性:时间与地点。《发育》146, dev182154 (2019)。
- 卢波(G.)、哈里斯(W. A.)和刘易斯(K. E.)。脊椎动物神经系统腹侧模式形成的机制。《自然综述·神经科学》7, 103–114 (2006)。
- 阿莫罗索(M. W.)等人。从人类干细胞快速高效生成支配肢体的运动神经元。《神经科学杂志》,第33卷,第574-586页(2013年)。
- 杜志文等人。从人多能干细胞中生成和扩增高纯度运动神经元祖细胞。《自然·通讯》6, 6626 (2015)。
- Rizzo, F. 等人。SMN/SYNCRIP和RNA基序7在脊髓性肌萎缩症中的关键作用:人类运动神经元的RNA测序和基序分析。《大脑》142,276-294(2019)。
- 张琦等人。单细胞分析揭示了人类脊髓发育中神经细胞的动态变化。《欧洲分子生物学组织报告》,22卷,e52728(2021年)。
- Rayon, T.、Maizels, R. J.、Barrington, C. 和 Briscoe, J. 对人类发育中的脊髓进行单细胞转录组分析,揭示了具有人类特异性特征的保守遗传程序。《发育》148,dev199711(2021)。
- Dann, E., Henderson, N. C., Teichmann, S. A., Morgan, M. D. 及 Marioni, J. C. 利用k近邻图对单细胞数据进行差异丰度测试。《自然·生物技术》40,245-253(2022)。
- Sagner, A. 等人。一套共享的转录密码协调中枢神经系统的时间模式形成。《公共科学图书馆·生物学》19, e3001450 (2021)。
- Van de Sande, B. 等人。用于单细胞基因调控网络分析的可扩展SCENIC工作流程。《自然·实验手册》15, 2247–2276 (2020)。
- 孟华等。帕金森病相关蛋白CHCHD2的缺失会影响线粒体嵴结构并使细胞色素c不稳定。《自然·通讯》8, 15500 (2017)。
- Kee, T. R. 等。线粒体CHCHD2:疾病相关突变、生理功能及当前动物模型。《衰老神经科学前沿》13, 660843 (2021)。
- 阿鲁穆加姆(S.)、加尔塞拉(A.)、索勒(R. M.)和塔巴雷斯(L.)。Smn缺陷会增加运动神经元的内在兴奋性。《细胞神经科学前沿》,11卷,269页(2017年)。
- Quinlan, K. A. 等。在脊髓性肌萎缩症的Smn2B/-小鼠模型中,运动神经元丢失之前出现过度兴奋。《神经生理学杂志》,122卷,1297-1311页(2019年)。
- Brunet, A., Stuart-Lopez, G., Burg, T., Scekic-Zahirovic, J. 及 Rouaux, C. 皮质回路功能障碍可能是肌萎缩侧索硬化症的一个潜在驱动因素。《神经科学前沿》14, 363 (2020)。
- Sibilla, S. 和 Ballerini, L. 脊髓发育过程中GABA能和甘氨酸能中间神经元的表达:抑制与兴奋在控制腹侧网络输出中的动态相互作用。《神经生物学进展》89, 46–60 (2009)。
- 拉米雷斯(A.)等人。新型吗啉代寡聚物提高脊髓性肌萎缩症患者存活运动神经元蛋白水平的研究。《国际分子科学杂志》,19,167(2018)。
- 莫尔顿,H. M. 和莫尔顿,J. D. 吗啉代及其肽偶联物:杜氏肌营养不良症的治疗前景与挑战。《生物化学与生物物理学报》1798,2296–2303(2010)。
- Bersani, M. 等人。细胞穿透肽偶联的吗啉寡核苷酸在有症状的临床前模型中挽救脊髓性肌萎缩症。《分子治疗》30, 1288–1299 (2022)。
- 古比茨(A. K.)、冯(W.)和德雷福斯(G.)。SMN复合体。《实验细胞研究》,296期,51-56页(2004年)。
- Yoo, H. 等人。ATP依赖的染色质重塑因子CHD9以细胞培养条件依赖的方式调控胚胎干细胞的增殖。《生物学》9, 428 (2020)。
- 郭伟等人。HDAC6抑制可逆转源自FUS-ALS患者的运动神经元中的轴突运输缺陷。《自然·通讯》8,861(2017)。
- 陈等人。组蛋白去乙酰化酶6通过改善肌萎缩侧索硬化症转基因小鼠模型中的自噬流缺陷来延缓运动神经元变性。《神经科学通报》,31卷,459-468页(2015年)。
- Mancinelli, S. 和 Lodato, S. 解码发育中大脑皮层的神经元多样性:从单细胞到功能网络。《当代神经生物学观点》53,146-155(2018)。
- 贝拉斯科(S. Velasco)、保尔森(B. Paulsen)和阿洛塔(P. Arlotta)。3D脑类器官:在胚胎外研究大脑发育与疾病。《神经科学年度评论》,43卷,375-389页(2020年)。
- Tambalo, M. 与 Lodato, S. 脑类器官:研究神经元回路组装、功能及功能障碍的人类3D模型。《脑研究》1746, 147028 (2020)。
- 保尔森(B. Paulsen)等人。自闭症相关基因集中于共享神经元类别的异步发育。《自然》602,268–273(2022)。
- Antón-Bolaños, N. 等人。脑嵌合类器官揭示个体对神经毒性触发因素的易感性。《自然》631, 142–149 (2024)。
- Mancinelli, S.、Bariselli, S. 和 Lodato, S. 人类脑类器官中电活动的出现。《干细胞报告》20, 102632 (2025)。
- Ogura, T., Sakaguchi, H., Miyamoto, S. 及 Takahashi, J. 从人类多能干细胞三维诱导背侧、中间和腹侧脊髓组织。《发育》145, dev162214 (2018)。
- 福斯蒂诺·马丁斯、J.-M. 等人。自组织三维人体躯干神经肌肉类器官。《细胞干细胞》26,172–186.e6(2020)。
- Grass, T. 等人。同源患者来源的类器官揭示脊髓性肌萎缩症发病早期的神经发育缺陷。《细胞报告·医学》5, 101659 (2024)。
- Galimberti, M. 等人。在镶嵌型端脑类器官中,健康细胞可非细胞自主性地挽救亨廷顿病的细胞表型。《自然·通讯》15, 6534 (2024)。
- Galgoczi, S. 等人。亨廷顿蛋白CAG扩增通过极性缺陷损害合成人类二维原肠胚的胚层模式形成。《发育》148, dev199513 (2021)。
- 法拉韦利(I.)和科尔蒂(S.)。脊髓性肌萎缩症——治疗时代的挑战。《自然综述·神经病学》,16,655-656(2020)。
- Motyl, A. A. L. 等。脊髓性肌萎缩症系统性发育异常的产前表现。《人类分子遗传学》,29,2674-2683(2020)。
- Tharaneetharan, A. 等人。脊髓性肌萎缩症小鼠小脑和运动皮层的功能异常。《神经科学》452期,78-97页(2021年)。
- Simon, C. M. 等人。运动回路的干细胞模型将运动神经元死亡与SMN缺乏诱导的过度兴奋分离开来。《细胞报告》16, 1416–1430 (2016)。
- Wainger, B. J. 等。肌萎缩侧索硬化症患者来源的运动神经元的内在膜兴奋性过高。《细胞报告》7, 1–11 (2014)。
- Burg, T. 等人。在肌萎缩侧索硬化症小鼠模型中,缺乏大脑皮层下投射神经元是有益的。《神经病学年鉴》88, 688–702 (2020)。
- 马奎斯(C.)、伯格(T.)、斯切基奇-扎希罗维奇(J.)、菲舍尔(M.)和鲁奥(C.)。在肌萎缩侧索硬化症的Sod1小鼠模型中,上下运动神经元变性在躯体定位上相关且在时间上有序。《脑科学》,11卷,369页(2021年)。
- Finkel, R. S. 等。诺西那生钠与安慰剂对照治疗婴儿型脊髓性肌萎缩症。《新英格兰医学杂志》,377卷,1723-1732页(2017年)。
- 段D.、戈曼斯N.、武田S.、默库里E.和阿尔茨马-鲁斯A. 杜氏肌营养不良症。《自然综述疾病引物》7, 1–19 (2021)。
- Corti, S. 等。脊髓性肌萎缩症患者的人诱导多能干细胞的基因校正。《科学转化医学》4, 165ra162 (2012)。
- Hensel, N. 等人。神经营养信号枢纽B-Raf的损伤导致脊髓性肌萎缩症中的运动神经元退化。《美国国家科学院院刊》118, e2007785118 (2021)。
- Taiana, M. 等人。对SMARD1患者来源细胞的分子分析表明,无义介导的mRNA衰变存在缺陷。《神经病学、神经外科学与精神病学杂志》93, 908–910 (2022)。
- Ronchi, D. 等人。SMN1 基因中新型剪接位点突变与极严重的 SMA-I 表型相关。《分子神经科学杂志》56, 212–215 (2015)。
- 帕利亚里(E.)等人。靶向STMN2促进脊髓性肌萎缩症的神经保护和神经肌肉恢复:来自脊髓性肌萎缩症体外和体内模型的证据。《细胞与分子生命科学》,82卷,29页(2024年)。
- 卡彭特(A. E.)等。CellProfiler:用于识别和量化细胞表型的图像分析软件。《基因组生物学》,7,R100(2006)。
- Schindelin, J. 等。Fiji:一个用于生物图像分析的开源平台。《自然方法》9,676-682(2012)。
- 蒙特威尔第(A.)、迪·多梅尼科(D.)、丹杰洛(E.)和马佩利(L.)。通过高密度多电极阵列记录揭示小脑回路中信号传播的各向异性和频率依赖性。《生物医学》11, 1475(2023)。
- Wolf, F. A., Angerer, P. 及 Theis, F. J.。SCANPY:大规模单细胞基因表达数据分析。《基因组生物学》,19,15(2018)。
- Wolock, S. L., Lopez, R. & Klein, A. M. Scrublet:单细胞转录组数据中细胞双峰的计算识别。《细胞系统》8, 281–291.e9 (2019)。
- Gayoso, A. 等人。一个用于单细胞组学数据概率分析的Python库。《自然·生物技术》40, 163–166 (2022)。
- Traag, V. A., Waltman, L. 与 van Eck, N. J. 从鲁汶到莱顿:确保社区连接良好。《科学报告》9, 5233 (2019)。
- Ahlmann-Eltze, C. 与 Huber, W. 单细胞RNA测序数据转换方法的比较。《自然-方法》20, 665–672 (2023)。
- Muzellec, B.、Teleńczuk, M.、Cabeli, V. 和 Andreux, M. PyDESeq2:一个用于批量RNA-seq差异表达分析的Python包。《生物信息学》39,btad547(2023)。
- Badia-i-Mompel, P. 等。decoupleR:从组学数据推断生物活性的计算方法集合。《生物信息学进展》,2,vbac016(2022)。
- Lange, M. 等人。用于定向单细胞命运图谱绘制的CellRank。《自然·方法》19, 159–170 (2022)。
- 古拉蒂(G. S.)等人。单细胞转录多样性是发育潜能的标志。《科学》367,405–411(2020)。
- Moerman, T. 等人。GRNBoost2 和 Arboreto:基因调控网络的高效且可扩展推断。《生物信息学》35,2159–2161(2019)。
- AUCell。Bioconductor http://bioconductor.org/packages/AUCell/。
- Setty, M. 等人。利用Palantir表征单细胞数据中的细胞命运概率。《自然-生物技术》,37卷,451-460页(2019年)。
- Faure, A. J. 等。绘制蛋白质结合域的能量和变构图谱。《自然》604, 175–183 (2022)。
- Petitpré, C. 等人。小鼠内耳发育的单细胞RNA测序分析揭示了听觉神经元多样化的分子逻辑。《自然·通讯》13, 3878 (2022)。
- DeTomaso, D. 和 Yosef, N. Hotspot识别单细胞基因组学多种模式中的信息基因模块。《细胞系统》12, 446–456.e9 (2021)。
- De Donno, C. 等人。单细胞数据集的群体水平整合实现跨样本多尺度分析。《自然·方法》20, 1683–1692 (2023)。
- Lotfollahi, M. 等人。通过迁移学习将单细胞数据映射到参考图谱。《自然-生物技术》40, 121–130 (2022)。
- 何 Z. 等人。人类神经类器官的综合转录组细胞图谱。《自然》635, 690–698 (2024)。
- Dobin, A. 等。STAR:超快速通用RNA-seq比对器。《生物信息学》29,15-21(2013)。
- Shen, S. 等。rMATS:从重复RNA-Seq数据中稳健且灵活地检测差异可变剪接。《美国国家科学院院刊》111,E5593–E5601(2014)。
- Zhou, Y. 等人。Metascape 为生物学家提供了一个面向系统级数据集分析的资源。《自然·通讯》10, 1523 (2019)。
¶ 致谢
作者感谢Associazione Centro Dino Ferrari的支持,以及Humanitas基因组学单元(HUGE)和Humanitas生物信息学单元在单细胞测序实验和分析方面提供的技术支持。我们感谢Teresa Rayon(英国剑桥Babraham研究所)分享人类胎儿pMN和MN细胞群的计数数据,感谢Hynek Wichterle(美国纽约哥伦比亚大学运动神经元中心)分享HB9抗体。我们感谢Dario Ronchi的宝贵意见和Stefania Sempreboni的技术支持。我们感谢Hong Moulton教授在吗啉代肽缀合方面所做的工作。
我们还感谢以下资助来源:
意大利卫生部资助项目RF-2018-12366357(S.C.)
意大利卫生部资助项目RF‑2016‑02362317(G.C.)
意大利卫生部,IRCCS基金会“Ca”Granda Ospedale Maggiore Policlinico,当前研究2026(230)(G. C.和S.C.)
意大利卫生部PNRR-POC-2023-12377653(S.C.)
Telethon基金会资助项目GGP14025(M.N.)
意大利卫生部5×1000医疗卫生研究(S.L.)
意大利卫生部拨款RF‑2018‑12365280(S.L.)
Cariplo Giovani 2019-1785(S. L.)
翁贝托·韦罗内西基金会(M. T.)
ERC启动基金IMPACT 101043003(S.L.)
最终搜索授权GR201912368561(S. L.)
本研究得到了“下一代欧盟”(NGEU)的支持,并由意大利大学与研究部(MUR)、国家恢复与韧性计划(NRRP)资助,项目名称为MNESYS(编号PE0000006)——“健康与疾病状态下神经系统研究的多尺度综合方法”(编号1553,2022年10月11日),资助对象为E.P.和E.D.。米兰大学病理生理学与移植系由意大利教育与研究部(MUR)资助:2023至2027年卓越部门计划。
¶ 作者贡献
S.C.、M.N.、S.L.、I.F.、M.T.和P.R.构思并设计了本研究项目。M.N.、P.R.、M.R.、G.F.、A.D.A.和L.C.开展了实验,并收集了关于诱导多能干细胞(iPSCs)生成和鉴定的数据。P.R.、I.F.和A.D.A.生成并鉴定了脊髓类器官。F.B.协助生成和鉴定脊髓类器官。M.T.、P.R.和I.F.生成并鉴定了脑类器官。S.M.和M.T.进行了图像采集,S.M.、M.T.和P.R.进行了图像量化。L.M.和E.D.在E.P.的帮助下进行了多电极阵列(MEA)记录和分析,C.C.与P.R.一同对TUNEL分析进行了成像和量化。I.F.和P.R.对SOX2和TUJ1计数的汇总数据进行了统计分析。C.P.、S.M.和M.T.开展了单细胞RNA测序实验,P.K.和I.S.进行了相关的计算分析。M.M.和E.M.P.分析了批量RNA测序和剪接数据。G.C.和S.P.为数据讨论以及对I.F.和P.R.的指导提供了帮助。I.F.、P.R.、M.T.、S.L.和S.C.撰写了手稿,所有作者都对稿件做出了重要贡献。S.L.和S.C.对本研究工作贡献均等。所有作者都对稿件的最终版本进行了严格审阅并予以批准。
利益冲突:作者声明不存在利益冲突。
¶ 补充信息
补充信息 在线版本包含可在以下网址获取的补充材料
https://doi.org/10.1038/s41467-025-67725-1。
通信及材料请求请联系Simona Lodato或Stefania Corti。
《自然·通讯》感谢池内义穂及其他匿名评审人员对本研究同行评审所做的贡献。同行评审文件已提供。
重印和许可信息请见http://www.nature.com/reprints
出版商说明:施普林格·自然对于已出版地图中的管辖权声明以及机构隶属关系保持中立。
开放获取 本文采用知识共享署名-非商业性使用-禁止演绎4.0国际许可协议授权。根据该协议,您可以以任何媒介或格式进行非商业性的使用、共享、分发和复制,但需适当注明原作者及出处,提供知识共享许可协议的链接,并说明是否对授权材料进行了修改。根据本许可协议,您无权共享改编自本文或其部分内容的改编材料。本文中的图片或其他第三方材料均包含在本文的知识共享许可协议中,除非在材料的引用说明中另有标注。如果材料未包含在本文的知识共享许可协议中,且您的预期使用未获得法律法规许可或超出许可范围,您需要直接从版权持有人处获得许可。如需查看本许可协议的副本,请访问http://creativecommons.org/licenses/by-nc-nd/4.0/。
⊚ 作者(们)2025年