¶ 显性β-连环蛋白突变导致具有可识别综合征特征的智力障碍
瓦尔特·图奇,1,2 蒂茨克·克莱夫斯特拉,3 安德烈亚·哈迪,2 伊内斯·海泽,2 西尔维娅·玛吉,1 马乔莱因·H·威廉森,3 海伦·希尔顿,2 克里斯·埃萨帕,2 米歇尔·西蒙,2 玛丽亚-特蕾莎·布埃纳维斯塔,4,5 利亚姆·J·麦格芬,4 露西·维佐尔,2 卢卡·多德罗,6 索蒂里奥斯·察夫塔里斯,6,7 罗萨里奥·罗梅罗,2 威利·N·尼勒森,3 利森卡·E·L·M·维瑟斯,3 马利斯·J·肯珀斯,3 安妮克·T·武尔托-范·希尔夫豪特,3 扎法尔·伊克巴尔,3 玛尔塔·奥兰多,1 亚历山德罗·马乔内,1 格伦达·拉西,1 帕斯夸利娜·法里塞洛,1 安德烈亚·孔泰斯塔比莱,1费德里科·蒂纳雷利,1 蒂埃里·尼厄斯,1 安德烈亚·拉伊蒙迪,1 芭芭拉·格雷科,1 丹妮拉·坎塔托雷,1 劳拉·加斯帕里尼,1 卢卡·贝尔东迪尼,1 安杰洛·比丰,6 亚历山德罗·贡齐,6 萨拉·韦尔斯,2 和 帕特里克·M·诺兰2
1.意大利技术研究院神经科学与脑技术系,意大利热那亚
2.MRC Harwell,哈维尔科学与创新园区,英国牛津郡
3.拉德堡德大学医学中心人类遗传学系,荷兰奈梅亨
4.雷丁大学生物科学学院,英国雷丁
5.钻石光源B23光束线,英国牛津迪德科特
6.意大利技术研究院神经科学与认知系统中心,意大利罗韦雷托
7.IMT高级研究学院卢卡,意大利卢卡。
近期在人类和小鼠中同时发现编码β-连环蛋白基因的多个显性突变,使得我们能够从分子和细胞层面探究β-连环蛋白在认知功能障碍中的作用机制。人类研究中已识别出导致一系列神经发育障碍的β-连环蛋白突变。我们通过在智力障碍患者中发现的全新β-连环蛋白突变,精细解析其表型特征,最终确定了可识别的智力障碍综合征。与此同时,对化学诱变小鼠品系(其表现与携带β-连环蛋白突变的人类患者特征相似)的研究,使我们能够追踪β-连环蛋白功能失调从发育期到成年期的全过程。这种被命名为batface(Bfc)的小鼠突变体,在β-连环蛋白C端犰狳重复序列中发生Thr653Lys替换,表现出与膜结合钙黏蛋白结合亲和力下降的特性。伴随这种钙黏蛋白相互作用的减弱,我们发现该突变导致大脑半球内连接减少,并引发树突分支缺损、长时程增强效应受损及认知功能障碍。本研究为β-连环蛋白显性突变导致其黏附功能丧失提供了体内证据,这种功能丧失会引发严重症状,包括智力缺陷、儿童期肌张力减退、进行性下肢痉挛以及成人颅面特征异常。
¶ 引言
-连环蛋白(CTNNB1)是一种高度保守的蛋白质,通过与细胞黏附蛋白、信号分子和转录因子相互作用来实现关键细胞功能(1)。 -连环蛋白的特征性结构——其12个中心犰狳重复序列——形成一个带正电荷的长沟槽,促进了与多种蛋白质伙伴的相互作用。该中心基序两侧分别为介导蛋白质降解至关重要的N末端,以及含有Helix-C基序(2)的C末端,该基序使蛋白质能够在细胞黏附与增殖的双重功能间进行切换。哺乳动物功能缺失研究显示β-连环蛋白参与胚胎发育,而功能获得研究则证明其与多种人类癌症的发生相关(综述见参考文献3)。功能研究主要聚焦于 -连环蛋白在经典WNT信号通路中的作用。 -连环蛋白通过与转录共激活因子相互作用来介导WNT的转录激活,这种激活作用在胚胎发育过程中协调生长与模式形成,而当其持续上调时,会导致与癌症和转移相关的生长失调。
除了在WNT信号通路中的作用外, -连环蛋白还作为膜钙黏蛋白的主要细胞内锚定物。钙黏蛋白是一类调节突触连接的细胞黏附分子(4)。其胞内结构域能够与 -连环蛋白的整个中央12臂重复基序建立相互作用。这种相互作用整合了多个独立过程(5),包括维持细胞黏附、调节细胞迁移及神经突向外生长(6, 7)。有模型提出β-连环蛋白通过调控这些相互作用参与突触重塑(8)。由于突触变化是记忆形成的关键细胞过程(9),钙黏蛋白-连环蛋白复合物的受控稳定性可能是记忆的细胞介质。事实上,通过条件性基因敲除实验已证实β-连环蛋白在长时程记忆巩固中发挥重要作用(10)。
通过高通量基因组学方法,近期有证据表明β-连环蛋白与人类认知障碍存在关联(11, 12)。大规模测序研究发现,CTNNB1上的罕见新生点突变是导致智力障碍(ID)(13)和自闭症谱系障碍(ASD)(12)的主要因素。这些研究在CTNNB1中发现了4个潜在致病突变:其中3个是ID患者的功能缺失突变(p.Ser425Thrfs*11、p.Arg515*和p )(13),另有1个是ASD患者的错义突变(p.Thr551Met)(12)。继我们先前的ID研究之后,本文对患者的表型特征进行了完整表征。此外,我们近期新发现一名携带从头突变CTNNB1(c.705dup;p.Gly236Argfs )的个体,此病例的突变数据与临床表现系首次披露。与此同时,我们鉴定出名为batface(Bfc)的小鼠模型,该模型因N-乙基-N-亚硝基脲诱导(ENU诱导)在β-连环蛋白中产生错义突变。Batface突变体表现出特殊的形态学、行为学、分子生物学及生理学异常,这些异常与同源人类病症存在显著对应人类疾病状况。我们收集并分析了人类患者及小鼠模型的表型数据,从而全面揭示 -连环蛋白在这些严重神经发育疾病的发病与进展过程中所起的作用。
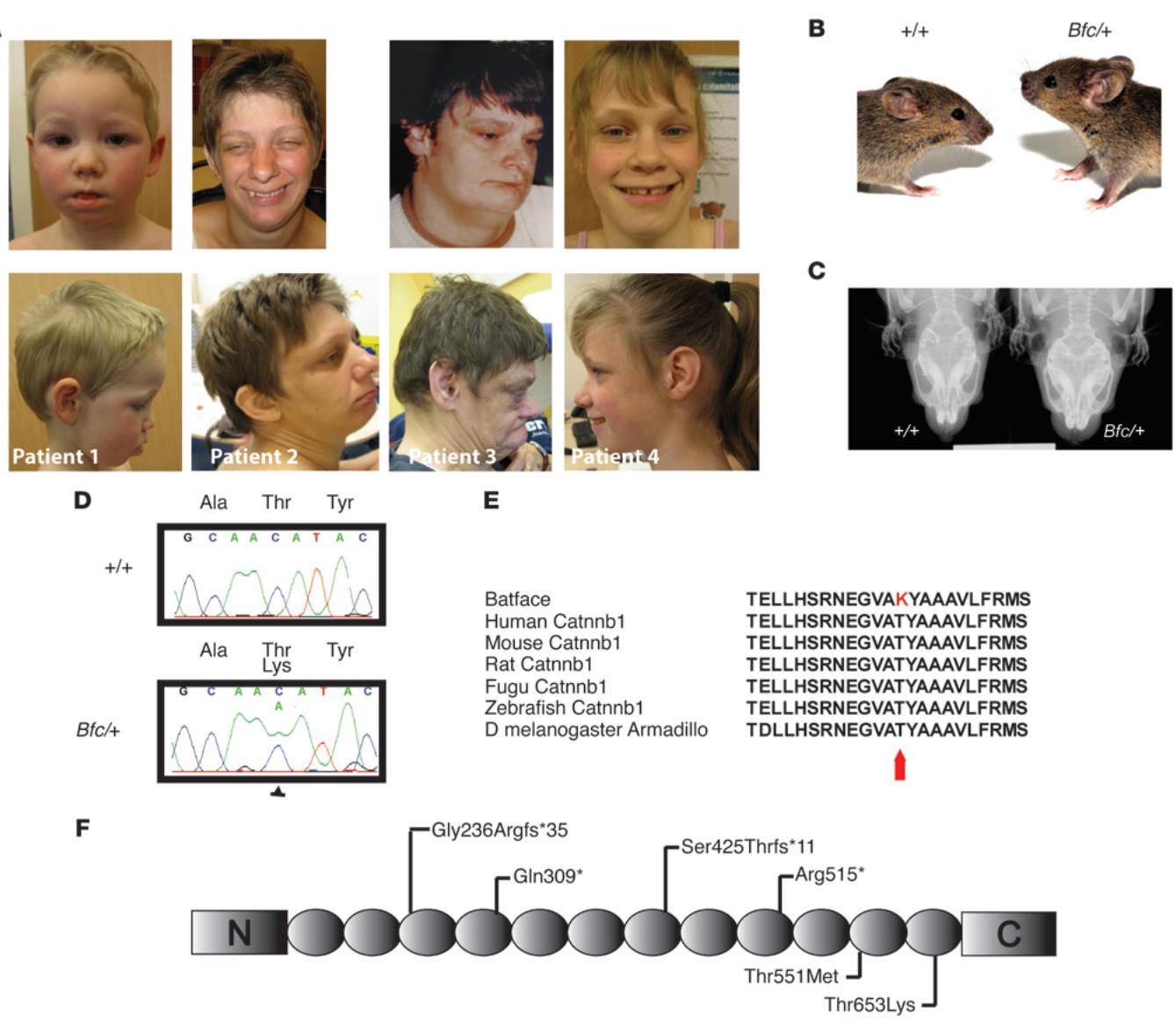
图1 人类和小鼠因β-连环蛋白突变引发的颅面异常。(A) 1号(4岁)、2号(29岁)、3号(24岁和51岁)及4号(14岁)患者携带CTNNB1新生突变(分别为p. 、p.Ser425Thrfs*11、 和p.Gly236Argf )。注意其共有的颅面特征包括小头畸形、鼻尖饱满及上唇偏薄。已获得4名患者法定代理人对使用医疗数据和照片的书面知情同意。(B) 所有Bfc/+成年鼠均表现出的宽脸表型特征(与WT同窝对照鼠相比)。© X射线扫描显示Bfc/+的宽脸表型与短鼻梁、宽头骨相关。(D) Bfc 与WT的Ctnnb1基因序列分析显示,Bfc突变体在2245核苷酸位点存在C到A的颠换,该错义突变导致Thr653Lys氨基酸替换。(E) 人类、小鼠及其他脊椎动物CTNNB1蛋白序列比对,Thr653Lys置换位于高度保守的第12个Armadillo重复序列内。(F) CTNNB1蛋白结构示意图显示N端与C端结构域及Arm12重复序列,标注了5个人类突变(包括文献12的Thr551Met)及小鼠Thr653Lys突变的大致位点。
¶ 结果
¶ 携带β-catenin基因新发突变的个体表现出一系列临床特征,这些特征与行为异常及神经发育障碍相关
基于最近在全基因组测序研究中反复出现的发现,促使我们 研究临床与人类新生β-连环蛋白突变相关的特征。我们对4名智力障碍个体进行了详细表型研究,并得出结论:他们表现出高度相似且独特的特征,促使我们定义了一种新型可识别综合征。该综合征包括轻度至重度智力障碍、自闭症谱系障碍、儿童期肌张力低下伴进行性痉挛性双瘫、(原发性)小头畸形,以及显著的颅面部和脑部异常(包括胼胝体发育不全)(图1A)。补充临床描述详见附录临床资料(在线补充材料见本文网址:doi:10.1172/JCI70372DS1)。由于所有已识别的智力障碍相关突变(图1F)均预测会导致功能丧失型蛋白截短,我们通过等位基因特异性cDNA扩增技术,在环己酰亚胺处理/未处理的EBV转化淋巴细胞中验证突变mRNA是否会发生无义介导的降解。在4名患者中有2人观察到该现象(补充图1)。这些结果表明CTNNB1单倍体不足是导致患者颅面部异常和智力障碍表型的原因。
¶ 一次乙基亚硝基脲(ENU)诱变筛选发现了一个β-连环蛋白小鼠突变体
在一项平行研究中,我们在ENU诱变动物(14)的G1群体中发现了一个个体,其表现出显性颅面表型,具有特征性的宽脸型特征(图1B和C)。该品系因其独特的颅面畸形被命名为batface(Bfc, MGI:2656734),其表型以显性方式遗传,而回交实验表明该突变具有纯合致死性,致死发生在妊娠中晚期(15)。我们使用13只患病动物对显性颅面表型进行了低分辨率定位。在发现远端9号染色体上存在单一连锁区域后,我们使用额外标记和动物将非重组区域精细定位至120.92 Mb至121.48 Mb之间的0.56 Mb区间。由于仅有2个转录本定位至该区域,我们对所有编码外显子进行测序,在β-连环蛋白基因(Ctnnb1;图1D)的外显子13中发现单个错义突变——C到A的转换。该突变导致蛋白C末端高度保守的第12个armadillo重复序列中发生Thr653Lys替换(图1E)。 妊娠中期胚胎的初步功能研究表明,Bfc突变通过WNT介导的转录通路中的功能获得性机制发挥作用(15)。但与其他β-连环蛋白的功能获得性或功能缺失性突变不同,据我们所知,这是首个小鼠突变实例指向杂合成年个体的颅面畸形。与同窝对照组相比,Ctnnb1Bfc/+ )小鼠存在显著颅骨差异,包括鼻长缩短、眶间距增宽以及前囟点到骨嵴距离增加(图1C和附图2)。考虑到人类CTNNB1突变与Bfc/+小鼠具有相似的表型特征,我们决定评估小鼠中Bfc突变的性质。因此我们开展了一系列蛋白质功能研究,以探究CTNNB1的任何显著功能是否在Bfc/+小鼠中受到影响。
¶ Bfc突变破坏了钙粘蛋白-β-连环蛋白的相互作用
为了确定Bfc突变的性质,我们首先检测了蛋白质水平是否受到影响(补充图3)。由于Western blot未发现差异,我们进一步探究该突变是否会影响β-连环蛋白与其已知相互作用因子的结合能力。具体而言,C端armadillo重复序列在锚定膜结合钙粘蛋白与细胞内细胞骨架的作用已得到充分证实(5, 16)。此外,与Bfc突变相邻的酪氨酸残基(Tyr654)对维持这种相互作用至关重要。N端 -螺旋结构的对接当Tyr654被磷酸化时, -连环蛋白第11-12个Armadillo重复序列形成的沟槽内与钙粘蛋白的结合力可降低 。为探究Thr653Lys是否同样会破坏N端钙粘蛋白螺旋的对接,我们从野生型和Bfc/+成年海马全细胞裂解液中免疫沉淀了天然蛋白。引人注目的是,我们发现Bfc 海马组织中钙粘蛋白-连环蛋白的相互作用显著降低了 ;t检验)(图2 A和B)。这种突变型β-连环蛋白与钙粘蛋白结合亲和力的降低,随后在转染N-钙粘蛋白和β-连环蛋白哺乳动物表达载体的HEK293细胞免疫沉淀物中得到证实(图2 C和D)。利用现有的β-连环蛋白与E-钙粘蛋白结构模型,我们研究了Thr653Lys替换是否可能破坏这两种蛋白质间的相互作用(图2 E-H及补充图4)。通过叠加突变型、野生型及Tyr654磷酸化β-连环蛋白的三维模型(图2H),观察到Tyr654磷酸化会破坏与钙粘蛋白Asp665的氢键(5)。在研究β-连环蛋白与钙粘蛋白的相互作用螺旋时发现,突变型β-连环蛋白的预测三维模型显示出与钙粘蛋白相互作用的破坏,导致空间位阻凸起,其效应与Tyr654磷酸化相似。
¶ 形态学分析揭示Bfc/+小鼠存在脑部异常
我们继续在神经解剖学和行为学水平上对Bfc进行表征。通过MRI成像,我们在突变体中发现了一系列重大异常。突变体的前后轴明显缩短,而背腹轴和内外侧轴则扩大(图3 A和B)。与过表达 -连环蛋白的小鼠研究结果(18)相反,我们未观察到大脑皮层的沟回形成。然而,Bfc/+的灰质和全脑体积显著更大( .05;单因素方差分析,补充图5A)。但当标准化为脑容积后,Bfc/+的相对灰质与白质含量与对照组无统计学差异(补充图5B)。我们还发现多个脑解剖结构的绝对体积和相对体积均存在改变(补充图6)。Bfc 的丘脑、纹状体和苍白球体积略有增大但具有显著性。然而与野生型相比,突变体的小脑和嗅球体积显著减小。除这些总体形态特征外,3个Bfc 个体的胼胝体似乎严重发育不良,缺乏任何半球间连接结构(图3 C-E)。这让人联想到前述人类胼胝体发育不全的病例。其余Bfc 脑组织或对照组中未观察到主要胼胝体改变的证据。通过小鼠脑组织病理切片证实了MRI检测到的嗅球缩小、小脑体积减小及胼胝体异常(补充图7)。
¶ Bfc突变小鼠表现出感觉运动门控、运动功能及发声复杂性方面的缺陷
我们研究了小鼠的特定行为,这些行为与人类智力障碍病例中明显的缺陷相关,包括运动功能障碍、认知能力缺陷和发声异常。听觉惊吓反应(ASR)(图4A)及其前脉冲抑制(PPI)(图4B)反映了动物对感觉信息作出反应和整合的能力(19)。Bfc/+小鼠表现出持续且显著的前脉冲抑制缺陷,而听觉惊吓反应正常。运动缺陷存在但更为细微。在典型的旋转棒测试中,Bfc/+小鼠未表现出显著缺陷(数据未显示)。然而,当采用更具挑战性的测试方案——在单次会话中连续进行6个加速阶段测试时,Bfc/+小鼠从棒上坠落的潜伏期显著缩短,且这种差异在多次重复进行3天(图4C)。小鼠在特定条件下会发出超声波叫声,例如母婴分离时。我们在出生后第6天通过将幼崽与母鼠分离5分钟来测试超声波发声。我们观察到突变体的总叫声次数显著减少(图4D)。每次叫声的平均持续时间略有缩短,但不显著。最后,突变体幼崽产生了更多以单一成分为特征的叫声(图4E和F),表明它们发育出了复杂度较低的发声系统。
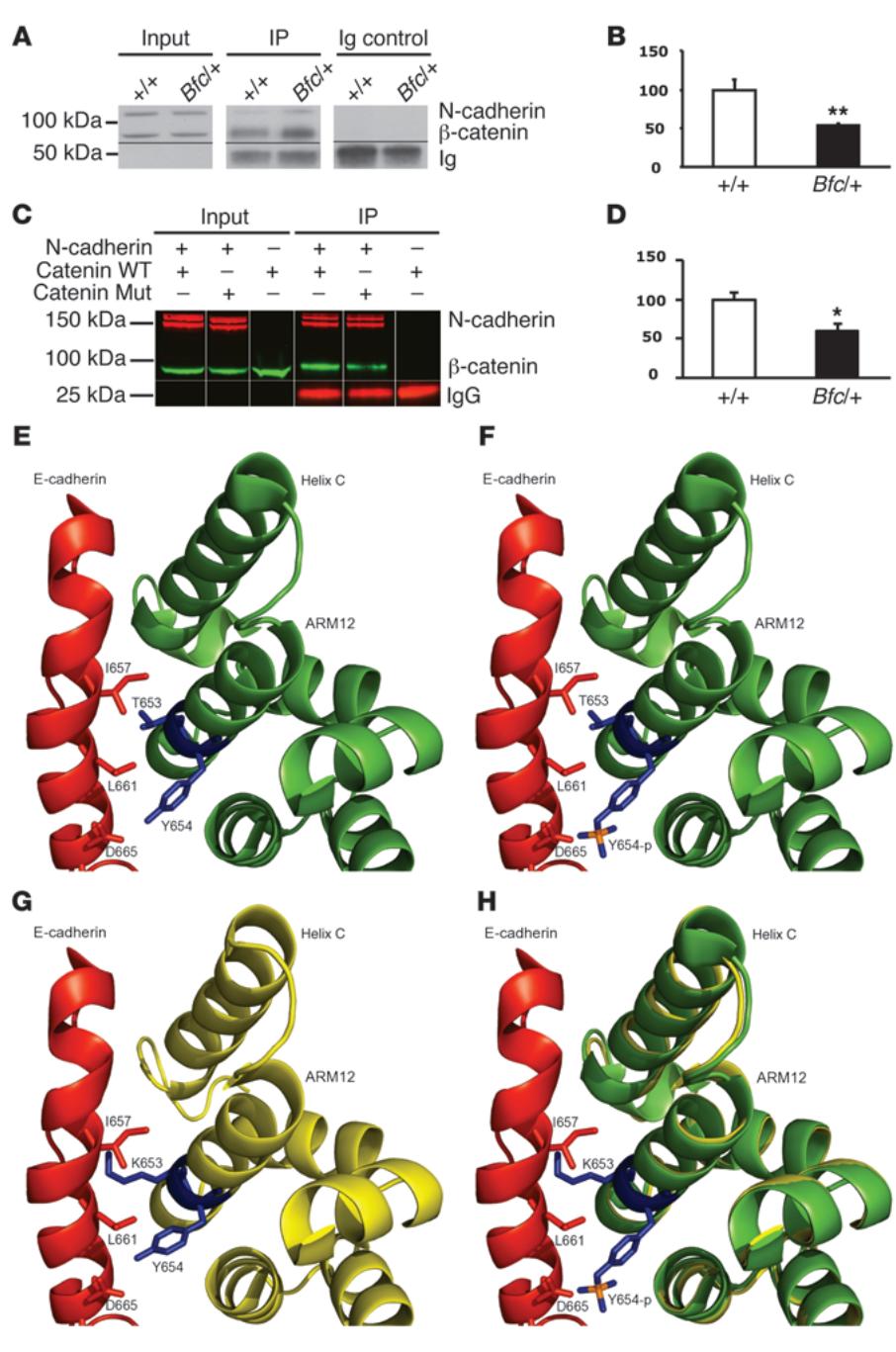
图 2 Thr653Lys突变破坏了小鼠β-连环蛋白-钙黏蛋白复合物。(A) 使用抗N-钙黏蛋白抗体对成年海马全细胞裂解液进行免疫沉淀的Western印迹结果。图中显示典型的N-钙黏蛋白和β-连环蛋白免疫反应条带。另设使用正常兔IgG的免疫球蛋白对照裂解液IP组。黑色线条标示非连续区域。(B) 将β-连环蛋白免疫反应性标准化至免疫沉淀的N-钙黏蛋白后,显示突变型海马裂解液中的相互作用降低 ( )。© 转染N-钙黏蛋白-EGFP和β-连环蛋白-V5/His的HEK293细胞裂解液经抗EGFP免疫沉淀的Western印迹。对照组IP仅转染W -连环蛋白-V5/His质粒。白色线条标示非连续区域。(D) 将重组β-连环蛋白免疫反应性标准化至免疫沉淀的重组N-钙黏蛋白,证实相互作用减弱(降低 , , )。(E)WT复合体中相互作用残基的特写视图。 -连环蛋白以绿色显示。 -连环蛋白中的T653和Y654(蓝色棒状结构)均位于ARM12螺旋3的表面。E-钙黏蛋白(红色)螺旋显示其接触残基D665和L661。E-钙黏蛋白的D665与Y654形成氢键。(F) 描述同E,但显示磷酸化酪氨酸残基(Y654-p)。(G) 突变型(T653K)相互作用残基的特写视图。基于IntFOLD2(35,36)模型的β-连环蛋白Armadillo结构域(黄色)显示K653(蓝色棒状结构)。该突变残基延伸至E-钙黏蛋白主链(红色)接触位点。(H) 将含K653突变的β-连环蛋白三维模型叠加于β-连环蛋白晶体结构,显示K653与Y654-p共同作用于E-钙黏蛋白相互作用残基:D665、L661和I657。
¶ Bfc突变体的学习与记忆缺陷
智力障碍个体的显著特征之一是认知缺陷。为研究Bfc/+小鼠的认知能力,我们采用了两项海马区相关测试:水迷宫测试(20)和恐惧条件反射测试(21)。在水迷宫测试(一项空间学习能力测试)中,我们对小鼠进行了为期4天的训练。
通过可见平台触发海马体非依赖性导航后,采用隐藏平台训练以评估海马体依赖性学习能力。两组小鼠在寻找可见平台前的潜伏期未见差异。然而,在隐藏平台测试中对照组表现持续提升,Bfc/+小鼠却未显现进步(图5A)。Bfc/+小鼠在每日训练结束后的探测试验中始终表现延迟(图5B),但其游泳能力正常(图5C、D),证实其海马体依赖性行为存在缺陷。在标准情境记忆恐惧 conditioning 测试中,Bfc/+小鼠再次暴露于情境时的僵直行为显著低于同窝对照组(图5C),表明突变体回忆先前电击经历的能力受损。
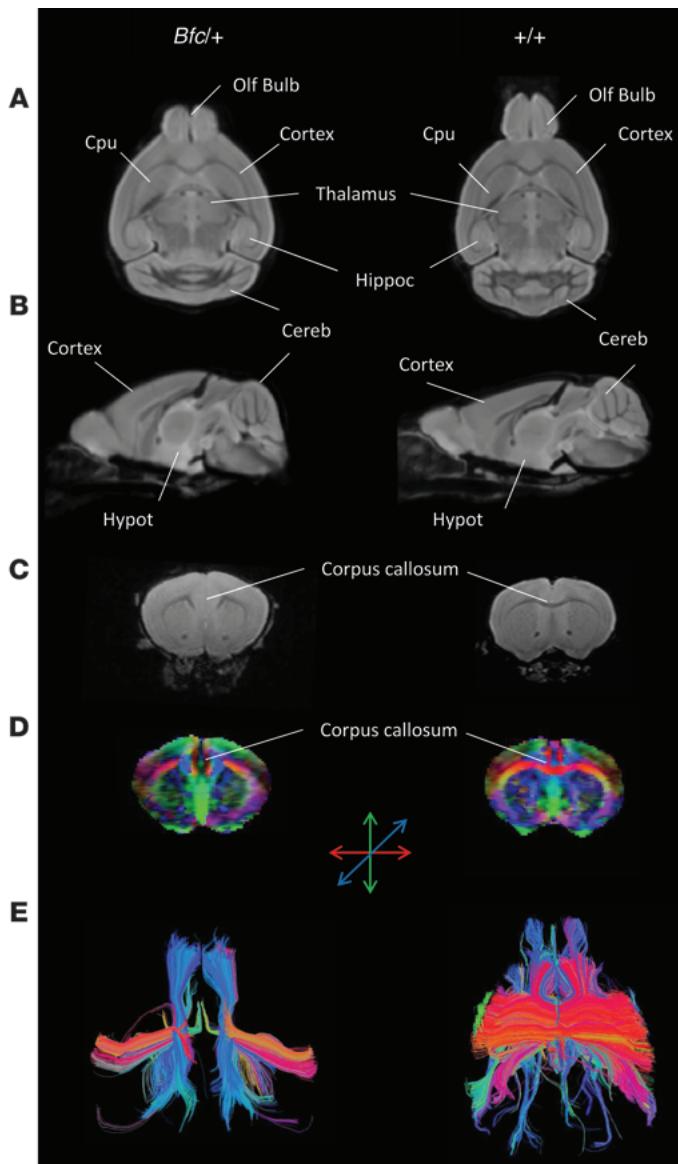
图 3Bfc 小鼠的MRI扫描显示其存在重大脑部异常。与对照组相比,Bfc/+小鼠的MRI扫描显示出颅骨形态改变——左右轴距增大且纵向尺寸缩短。这一现象在脑部水平切面(A)与矢状切面(B)中均尤为明显。Bfc 个体的嗅球与小脑体积较同窝对照组显著缩小。在10只Bfc/+实验对象中,有3只表现为胼胝体严重发育不全,完全缺乏半球间延伸结构。该缺陷在解剖T2加权像(C)及弥散加权扫描(D与E)中均得到印证。经FA调制的弥散张量成像(D)与DTI纤维束追踪共同证实,这3只实验对象均存在胼胝体半球间连接缺失,解剖MRI图像(C、D、E展示了典型Bfc/+与对照组数据)均显示胼胝体解剖结构异常,而所有对照组均观察到正常的半球间神经纤维束。缩写说明:Cereb:小脑;Cpu:尾状壳核;Olf Bulb:嗅球;Hippoc:海马体;Hypot:下丘脑。
¶ Bfc突变体中时序与决策过程发生紊乱
为测试认知处理的时间特性,我们采用基础"峰值程序"范式(22):训练小鼠在光信号事件后的特定时间间隔获取奖励(图6A)。在给予奖励的试验中,野生型小鼠在预期时间区间内反应活动显著增强,而Bfc/+突变体则无此现象(图6B)。此外,在未设置奖励的探测试验中,Bfc/+小鼠在关键时间窗内的反应显著低于同窝对照组(图6B)。通过自动化居家笼舍版峰值程序方案(23),我们连续多日记录小鼠正常睡眠-觉醒周期中的行为表现。这种更精确的时间反应分析证实了学习缺陷的存在。发现Bfc/+小鼠训练首日与末日的峰值分布无差异,而对照组则缩短了峰值时间并收窄了时间反应的分布范围(图6C、D)。我们通过鼻触反应累积分布曲线(附图8)提取"起始"与"终止"指数,分别评估预期行为与持续性行为。值得注意的是,在训练过程中Bfc/+小鼠未能调节其反应次数,表明其缺乏时间协调性和刻板行为(图 6E)。此外,我们通过让小鼠在对应位置进行鼻触来测试它们能否区分短间隔与长间隔光信号。该测试(24)用于评估动物是否具有准确的内源性(信号持续时间的主观估计)和外源性(长短信号比率)时间表征。在12小时光照/12小时黑暗循环的暗期(即小鼠最活跃的阶段),Bfc/+小鼠的准确度和精密度显著下降(补充图9,A和B)。通过对错误率(即在错误位置进行鼻触的次数)的分析进一步证实了这一点。对照组小鼠的错误率仅在光期升高,而Bfc 小鼠始终维持较高错误率(图 6,F和G)。
¶ Bfc小鼠的突触形态和功能发生改变
我们研究了行为缺陷是否可能与Bfc/+大脑神经元结构和功能连接异常有关。首先观察了胚胎海马原代培养神经元的神经突生长和分支情况。发现在培养仅1天后,杂合子与纯合子Bfc神经元的形态已出现差异(补充图10A)。特别值得注意的是,Bfc突变体的神经突长度和数量显著增加(补充图10B)。然而培养8天后,大多数纯合子神经元失去结构完整性,同时Bfc/+神经元的树突分支程度显著低于对照组(图7 A和B)。此外,高钾+对树突分支和神经突长度的去极化效应在Bfc/+神经元中受到抑制(补充图11)。鉴于已发现的人类新生β-连环蛋白突变会导致mRNA发生无义介导的衰变,进而引起CTNNB1单倍体不足,我们研究了β-连环蛋白表达降低对神经元发育的影响。为此,我们使用针对CTNNB1的siRNA转染来自野生型小鼠的原代神经元培养体系。该siRNA使野生型培养体系中β-连环蛋白表达降低 (图7D),从而很好地模拟了携带β-连环蛋白突变患者体内的表达下降情况。与在Bfc突变小鼠神经元培养物中观察到的神经突延伸减少相一致(图7B),在野生型培养体系中进行siRNA介导的β-连环蛋白敲低后,与转染对照siRNA的培养体系相比,神经突长度和突起数量均显著减少(图7D),这表明β-连环蛋白在塑造神经元结构方面起着关键作用。
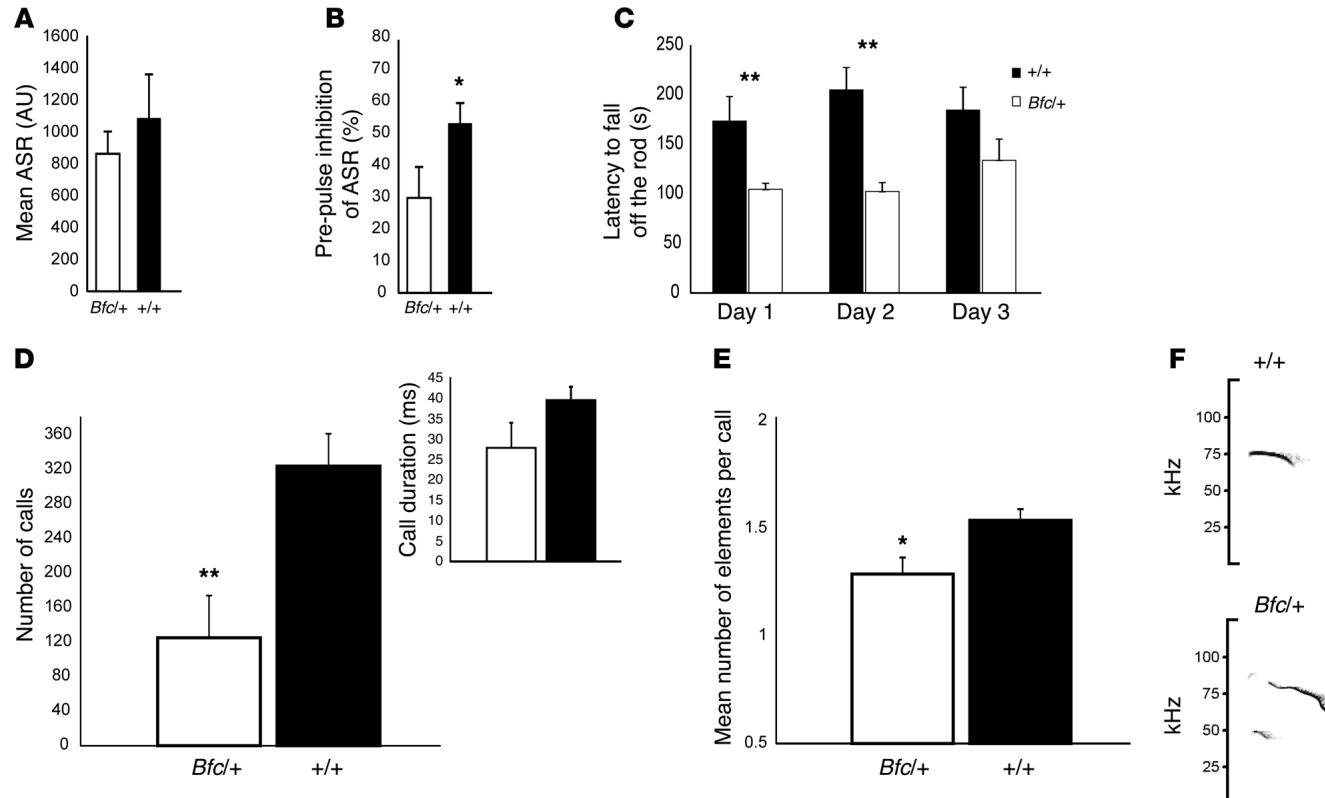
图4 成年小鼠的行为表型。(A) 与对照组 )相比, 小鼠 的听觉惊跳反应未受影响。(B) Bfc 小鼠的前脉冲抑制显著降低,且这种减弱效应极为显著。© 在连续3天的旋转棒测试中,Bfc 小鼠 )的表现虽在第三天有所改善,但整体差于对照组 。(D) 与同窝对照组 )相比, 小鼠 )的超声发声能力受损,表现为分离时的总叫声次数减少及平均叫声持续时间缩短。(E) 突变体的叫声复杂度也较低,每次呼叫包含的音节数量显著减少。(F) 野生型与Bfc 同窝小鼠的代表性超声发声图谱。 , ,学生t检验。
为探究结构变化是否会导致神经元网络尺度的功能差异,我们在高密度多电极阵列上培养海马神经元。该电极阵列支持在2.67平方毫米2区域内通过多达4,096个电极记录细胞外动作电位。为识别不同培养网络的功能差异,采用低密度细胞接种(图7E)形成稀疏神经网络,结合大规模电极阵列实现了单单元记录电极的高占比。在此条件下,峰值分类结果显示约84%的选定电极可记录单单元(附表1)。我们分别记录了自发状态及使用荷包牡丹碱解除抑制后的电生理活动。全网络活动参数(平均放电率与平均爆发率)的累积分布曲线均向右偏移,表明Bfc/ 小鼠神经元网络具有更高兴奋性。平均爆发时长的分析差异较不明显。这种高兴奋性在分别分析自发与解除抑制数据集时同样存在(数据未显示)。此外,基于网络中大量单单元间功能连接性评估结果,我们进一步评估了突触强度。通过分析培养网络内最强连接的长度(图7I),观察到Bfc/+神经元网络相较对照组能维持更长的连接距离。连接强度与观察到Bfc 网络的连接程度较低(数据未显示)。这表明来自Bfc 杂合子小鼠形成的海马神经元网络中的功能连接,尤其是在局部回路水平上,效率较低。
我们探究培养神经元网络的功能差异是否与成年Bfc/+小鼠大脑的超微结构异常有关。对成年大脑切片皮质及CA1海马区进行电子显微镜分析发现,杂合子小鼠皮质和CA1区突触前膜停靠囊泡密度较低(图8A、B)。这种囊泡密度降低可能意味着Bfc/+突触的释放概率下降,从而导致突触强度减弱。为深入验证,我们通过刺激谢弗侧支通路记录海马切片CA1区场兴奋性突触后电位(图8C),发现Bfc 的fEPSP斜率与对照组无显著差异(图8D,双因素方差分析, ),配对脉冲比率也无统计学差异(图8E,双因素方差分析, )。进一步研究突变对突触可塑性的影响发现,无论是强直刺激(图8F)还是θ簇刺激(图8G)诱导的长时程增强,在 小鼠脑切片中均显著减弱。

图 小鼠多项学习记忆参数存在缺陷。工作记忆测试显示,在8天训练期间(A)和探测试验(B)中,Bfc 小鼠及其同窝对照鼠( )找到逃生平台的平均潜伏期如图所示。训练过程中标明了可见平台(VP)与隐藏平台(HP)两种条件。常规训练(C)与探测试验(D)的平均移动距离如图所示。在恐惧条件反射测试中,展示了 小鼠( )与同窝对照鼠( )在适应训练阶段(E)以及情境/线索暴露阶段(F)的僵直时间。 , ;学生t检验。
在小鼠和人类中均得到验证。这印证了近期提出的观点: -连环蛋白相互作用网络在异常神经发育病因学中具有重要作用(12, 25)。我们得以在人类中界定出一种具有临床识别度的综合征,其特征表型包括智力残疾、自闭症谱系障碍、小头畸形及儿童期肌张力低下伴进行性痉挛性双瘫,这些表型与CTNNB1的新生突变相关。在自闭症谱系障碍研究(12)中报道的CTNNB1基因p.Thr551Met突变个体可能也符合这一临床谱系,因为该病例除自闭症谱系障碍外,还被描述具有中度智力残疾范围内的非言语智商(NVIQ 58)。
然而,临床数据确实被提供了。有趣的是,我们的研究结果也与另一发现相吻合:在自闭症谱系障碍患者尸检脑组织的前额叶皮层中,Wnt蛋白和β-连环蛋白的表达水平有所降低。这些缺陷与树突分支异常及星形胶质细胞胞体缩小相关(26)。此外,该表型特征与蝙蝠脸小鼠模型的高度相似性,强烈提示人类与小鼠在形态学和神经发育过程中存在共同的病理机制。因此,我们利用蝙蝠脸小鼠模型进一步探究了 -连环蛋白在中枢神经系统功能中的作用,并已确定了可能导致一系列发育、行为和认知缺陷的具体分子及突触缺陷。
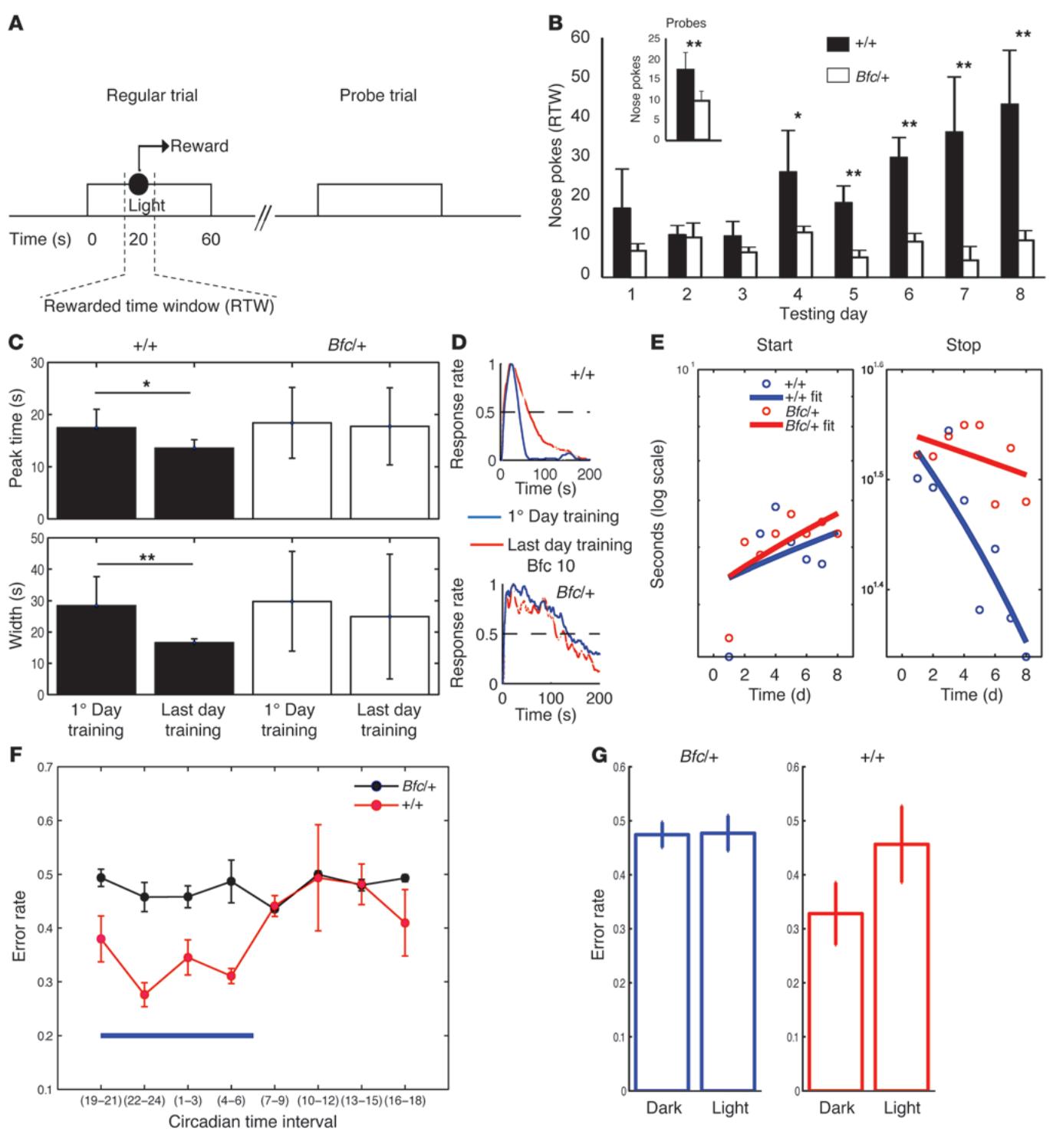
图6 Bfc 小鼠表现出时间认知缺陷。(A)峰值程序任务。探测试验与常规试验不同,光线刺激后不会伴随任何奖励。(B)展示了连续8天常规试验及所有探测试验中,各会话内奖励时间窗口的平均鼻触次数。Bfc 小鼠 未能像 小鼠 )那样在奖励时间窗口内增加鼻触行为。©通过独立小鼠队列 )在居家笼装置中收集时间反应。 组小鼠的峰值时间和宽度(离散度)在首末测试日间显著下降,而Bfc 组无此变化。*P ; 。(D)代表性 $: + / + $ 与Bfc 小鼠首日(红色)和末日的峰值分布。(E)起始与终止时间测试分别反映预期性和持续性行为。野生型小鼠表现出与奖励发放时间相匹配的反应优化(起始时间推迟、终止时间提前)。值得注意的是,Bfc 小鼠未调整终止时间,表明其在试验中存在持续性反应。(F)针对长短两个时间间隔进行评估,仅正确反应可获得奖励。错误率按3小时区间统计绘图;蓝色条表示12小时明暗循环中的暗相。通过对比Bfc 与 小鼠在暗相(活动期)与明相(静止期)的行为差异(G)发现:Bfc/+小鼠无相位行为差异,而野生型动物在明相错误率显著升高。
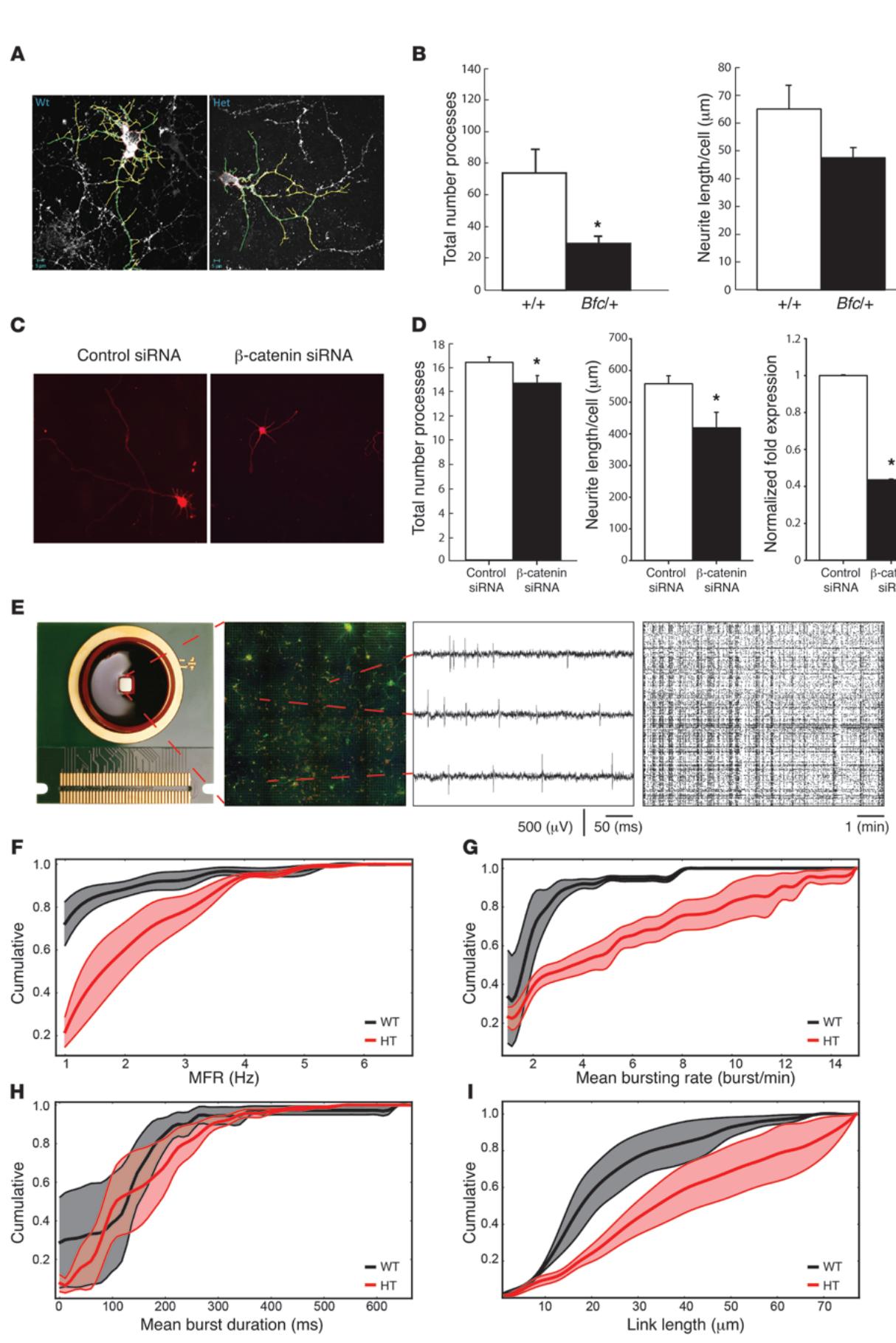
图7 Bfc 原代海马神经元培养物的形态和功能缺陷。(A)培养8天后 与Bfc/+海马神经元原代培养的代表性图像。比例尺如图所示。初级突起呈绿色,次级突起呈黄色。(B)呈现突起数量和神经突长度。Bfc/ 神经元的突起显著减少。 ,双尾t检验。(C)在1 DIV时转染β-连环蛋白或对照siRNA,7 DIV时固定的神经元培养物代表性图像。(D)RT-qPCR评估显示,针对 -连环蛋白的siRNA有效降低了mRNA表达( ),而转染β-连环蛋白的神经元中神经突延伸显著减少( )。(E)体外神经元网络设置示意图(从左至右):高密度MEA芯片、低密度培养荧光图像( -微管蛋白绿色,NeuN红色)显示电极阵列部分区域(黑色方框)的稀疏网络分布、3个不同通道采集的锋电位活动原始数据轨迹、约400个活跃通道(即发放频率> 0.1 spikes/s)的光栅图显示同步发放和持续簇状放电活动。(F–I)网络参数累积分布图( 黑色, ;Bfc/+红色, )。粗线与阴影区域表示均值 。Bfc 培养物兴奋性更高,表现为更高的MFR(F)和MBR(G)。MBD变化较不明显(H)。基于互相关的功能连接分析显示Bfc/+的功能连接长度也更高(I)。
蝙蝠脸突变体表现出一种独特的表型,这不仅是WNT相关功能获得(15)的结果,正如我们在此证明的,也是对黏附相关功能产生显性负效应的共同后果。我们相信这首次实现了在成年杂合子中研究突变体β-连环蛋白功能的体内效应。虽然条件性缺失突变体已发现特异性胚胎脑缺陷(包括部分病例中胼胝体发育异常)(27-29),但这些研究均未聚焦于出生后突变表型,也未在杂合子中观察到形态异常或行为缺陷。尽管杂合性功能缺失小鼠突变体未出现缺陷的情况,与人类β-连环蛋白功能缺失相关的严重表型似乎不一致,但Huelsken等人(30)提出斑珠蛋白在缺失状态下可承担β-连环蛋白的黏附功能。这不仅可解释小鼠与人类功能缺失突变的表现差异,也可能揭示Bfc突变独特性质的成因。鉴于突变体β-连环蛋白仍具有与细胞骨架及其他蛋白质伙伴相互作用的能力,Bfc突变蛋白的存在很可能抑制斑珠蛋白在杂合子动物中发挥其代偿性黏附功能的能力
作为CTNNB1及其相互作用因子复杂网络的印证,蝙蝠脸突变的严重程度及其多效性会随遗传背景的不同而产生差异。在本研究及既往研究(15)涉及的所有同类遗传背景中,虽然颅面畸形和某些行为特征表现稳定且不受遗传背景影响(数据未显示),但杂合子胚胎的存活率和成体生育能力均出现下降。为获得足够数量的研究动物群体,我们选择采用混合遗传背景的动物。这虽未影响行为学参数分析,但确实在少数动物中观察到低外显率胼胝体异常的出现。
就其具体的分子病变而言,与本文所述突变最为接近的是靶向磷酸模拟突变(Tyr-654Glu)(31)。Murase等人(8)提供了关键证据,表明Tyr654磷酸化状态对协调活动诱导的突触前和突触后可塑性至关重要。由于Bfc突变的效果似乎模拟了Tyr654磷酸化,我们的突触形态和功能结果与预期在突触连接处和网络层面出现的特异性异常相一致。Bfc与Tyr654Glu突变也存在诸多表型相似性,包括纯合子中的前段躯干截短和晚期胚胎致死性,以及与钙黏蛋白相互作用减弱。然而Tyr654Glu突变在WNT信号通路中表现出更强功能获得性,这一点在比较成年杂合子表型时尤为显著——与Bfc/ 小鼠不同,Tyr654杂合子未见颅面表型记录,但显示肠道腺瘤的肿瘤发生增加。
Bfc与Tyr654Glu突变体之间的差异可能与这两种突变体进行核输入和输出的能力不同有关,这些过程也对Tyr654磷酸化状态特别敏感(32)。类似地,Arm12中的氨基酸取代会影响该重复序列与 -catenin的C端结构域相互作用的能力。螺旋C可向后折叠至Arm12上,阻断转录共激活因子与该区域的结合,而Tyr654的磷酸化会阻止这种折叠,导致Ser675的后续磷酸化及转录共激活因子(包括CBP和TBP)的募集(33)。Y654E突变体确实表现出对Ser675磷酸化的敏感性增强;然而,虽然我们无法确定Bfc/+小鼠中Ser675的磷酸化状态,但未检测到突变蛋白与转录共激活因子CBP相互作用能力有任何变化(数据未显示)。这两种突变蛋白功能的细微差异可能是观察到的离散表型结果的基础。
培养的Bfc海马神经元中的缺陷与过表达β-连环蛋白的研究结果一致。稳定化β-连环蛋白的表达可促进树突生长和分支形成,但这一效应会被共表达的N-钙黏蛋白胞内结构域所抑制,该结构域能够隔离β-连环蛋白(34)。此外,高钾+对树突分支化和神经突长度的去极化效应,可通过过表达显性阴性形式的N-钙黏蛋白或存在Bfc等位基因而被抑制。关键的是,这些效应已被证明与Wnt相关的转录变化无关,尽管Wnt诱导的β-连环蛋白稳定化可能在介导去极化诱导的变化中具有重要作用。
钙粘蛋白/β-连环蛋白复合物先前已被证明在突触处突触小泡(SV)的募集和定位中发挥作用(8),而β-连环蛋白对于控制囊泡簇的大小和定位至关重要。事实上,海马锥体神经元中 -连环蛋白的缺失与储备池囊泡密度的降低有关。我们已证明,相对于野生型分离网络,Bfc/+的整体功能连接性较弱。特别是神经元之间的局部连接似乎受损最严重,这可能是由低效突触所致,从而损害了信号传输的可靠性。这种效率损失解释了为何Bfc 网络在单次放电和簇状放电活动中的活跃度均更高。
我们的研究结论支持β-连环蛋白/钙粘蛋白相互作用在调控突触可塑性与神经元网络连接性的分子机制中发挥核心作用。我们设想我们的遗传模型能够帮助深入理解分子机制,从而揭示β-连环蛋白在调控突触变化及最终影响认知功能中的作用。此外,通过该小鼠模型探究β-连环蛋白功能的能力,也将有助于我们理解并可能治疗一系列与神经发育缺陷相关的人类疾病,包括智力障碍和自闭症谱系障碍。
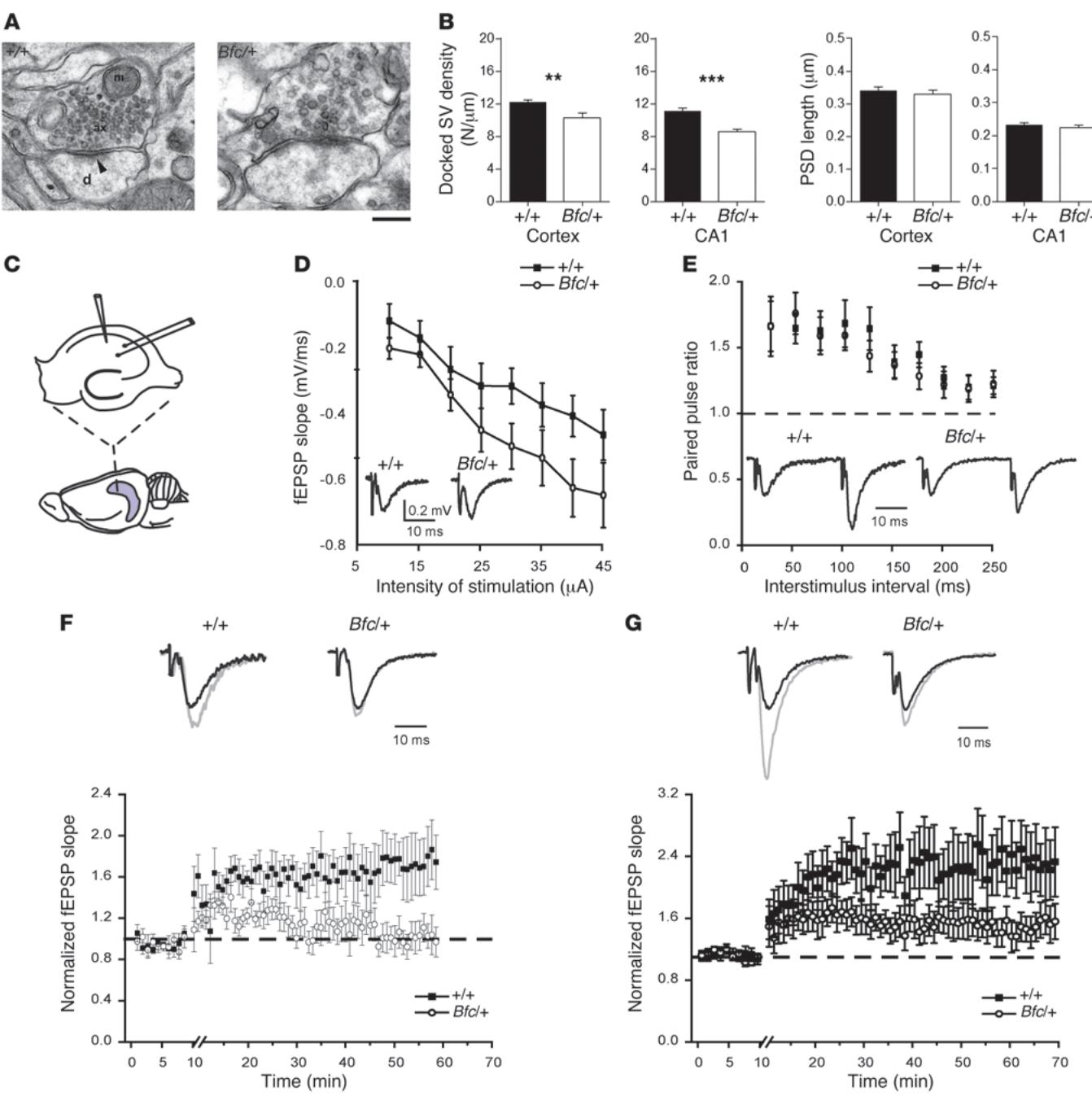
图 8 Bfc 成年海马体的形态和功能性突触缺陷。(A) 野生型和杂合子CA1突触的代表性电镜图像:显示包含突触小泡簇和线粒体(M)的突触前轴突终末(ax),以及突触后神经元树突(D);突触后致密区(PSD)亦可见(箭头标示)。标尺:200纳米。(B) 使用ImageJ对142个CA1和128个皮质 兴奋性突触前终末 )及152个CA1和115个皮质Bfc 兴奋性突触前终末 )进行膜锚定突触小泡密度(小泡数 米)和PSD长度(μ米)定量分析。©在成年海马切片中进行电生理记录的实验配置。(D) 野生型(黑色方块, 只小鼠,10切片)和Bfc/+(白色圆圈, 只小鼠,10切片)海马切片中fEPSP斜率与注入电流的函数关系。插图显示代表性波形。(E) 野生型 只小鼠,9切片)和Bfc 只小鼠,9切片)小鼠配对脉冲比率与刺激间隔的函数关系。插图波形均一化至首刺激fEPSP振幅。(F和G) 强直刺激(F)或θ串刺激(G)前后野生型(黑色方块)与Bfc/+(白色圆圈)切片中fEPSP斜率变化。数值均一化至对照组fEPSP斜率值。(F) 野生型 只小鼠5切片;Bfc/+ 只小鼠6切片。(G) 野生型 只小鼠5切片;Bfc/+ 只小鼠6切片。插图波形均一化至对照组fEPSP振幅。 , 。
¶ 方法
¶ 患者招募
当前这4名个体因中重度智力障碍被转诊至拉德堡德大学医学中心人类遗传学系进行评估。所有患者此前均已完成临床评估,并依据指征进行了特异性DNA检测及SNP芯片分析,结果均显示正常。随后,其中2名患者(患者2和4)被纳入家系全外显子组测序研究(三重分析)。患者2的研究结果已由本团队近期发表(三重分析70号,de Ligt等人;参考文献13)。患者4的结果此前未曾报道。患者1和3则被纳入包含765名智力障碍患者的验证队列,我们采用高通量重测序技术筛查包括CTNNB1在内的新候选基因,如本团队同期论文所述(分别作为第3位和第2位携带CTNNB1突变的患者,补充图7及de Ligt等人;参考文献13)。
¶ 动物
使用TaqMan等位基因判别分析法在标准条件下对动物进行基因分型。所用引物如下: -TGGCCTCAAGGGC-TAGTTTCT(正向)和5′-TGAAAGCCGCTTCTTGTAATC(反向)。TaqMan MGB探针序列为:FAM-TTTATTCCAGCAACATAC(野生型)与VIC-TTTTATTCCAGCAAAATA(突变型)。为维持Bfc突变品系的生育能力,通过将近交C57BL/6J背景的杂合子与C3H/HeH小鼠杂交进行繁殖。随后将突变型F1代与野生型F1代交配以构建实验队列。除非特别说明,本研究所有表型分析及组织来源均使用独立队列。行为测试后处死动物,发现大体解剖结构异常的个体均被排除研究。采用pDEXA Sabre X射线骨密度仪(诺兰医疗系统)进行X射线检测:简要流程为麻醉小鼠并置于检测台,以40毫米/秒的扫描速度和 毫米/秒的分辨率(HAV:0.020)进行扫描,骨参数测量通过配套软件完成。
¶ 细胞转染、蛋白质免疫沉淀、电泳和蛋白质印迹
HEK293T细胞通过jetPEI试剂(Source Bioscience)共转染N-钙黏蛋白–EGFP(质粒#18870;Addgene)与WT– -连环蛋白–V5/His或Bfc– -连环蛋白–V5/His(由澳大利亚国立大学的R. Arkell和A. D‘Cruz馈赠)全长cDNA构建体(各 ),培养48小时。通过RIPA缓冲液裂解制备培养细胞提取物( 每组3个)或全海马组织样本( 每种基因型7个),方法如前所述(37)。使用Dynabeads–Protein G免疫沉淀试剂盒(Invitrogen)从全海马提取物中免疫沉淀N-钙黏蛋白/β-连环蛋白复合物,操作遵循制造商说明。免疫沉淀使用 裂解液,洗脱复合物的 上样于聚丙烯酰胺凝胶。参考组同时上样 总裂解液。采用Invitrogen系统进行电泳后,通过iBlot(Life Technologies)转印至硝酸纤维素膜。膜在封闭液(含 脱脂奶粉的PBS Tween 20)中封闭1小时,随后在 条件下与封闭液稀释的一抗共同孵育过夜。经PBS含 Tween 20充分洗涤后,膜与封闭液稀释的相应二抗孵育1小时并重复洗涤。针对蛋白稳定性与免疫沉淀研究,使用β-连环蛋白C末端抗体(ab16051;Abcam)及N-钙黏蛋白抗体(ab18203;Abcam)。对照免疫沉淀采用正常兔IgG(sc-2027;Santa Cruz Biotechnology Inc.)。所用二抗为羊抗兔IgG-HRP(A6154;Sigma-Aldrich)和羊抗鼠IgG-HRP(A4416;Sigma-Aldrich)。转染细胞免疫沉淀中,裂解液与抗GFP单克隆抗体(Roche)在 孵育过夜,随后与蛋白G–琼脂糖珠(P3296;Sigma-Aldrich)在 共孵育1小时,用RIPA缓冲液洗涤珠子3次。对照实验在未转染N-钙黏蛋白条件下进行。通过LDS缓冲液(Life Technologies)煮沸5分钟从珠上洗脱免疫沉淀蛋白。定量双色Western印迹分析中,膜经抗GFP(Santa Cruz Biotechnology Inc.)和抗V5(Life Technologies)抗体探针孵育、洗涤后,与IRDye 800和IRDye 700红外荧光染料标记二抗共同孵育,方法如前所述(37)。使用Odyssey红外成像系统(LI-COR Biosciences)进行同步双色检测。免疫沉淀物中β-连环蛋白–V5的定量通过Odyssey成像软件分析,并以N-钙黏蛋白–EGFP荧光值标准化。
¶ β-catenin与E-cadherin的三维建模
-连环蛋白/E-钙黏蛋白复合物晶体结构的原子坐标(PDB 1I7X)(5)以及β-连环蛋白全长晶体结构(PDB 2z6h)(2)均从蛋白质数据库下载,并使用PyMOL进行可视化呈现。通过IntFOLD2折叠识别方法(35,36),我们针对在Arm12结构域发生Thr653Lys点突变的β-连环蛋白序列构建了全原子三维模型。利用PyMOL中的Builder工具,对β-连环蛋白野生型结构中磷酸化酪氨酸侧链(Tyr654-p)进行了建模。将2Z6H的β-连环蛋白晶体结构与1I7X中原始β-连环蛋白链进行叠合,展示了与E-钙黏蛋白结构结合的全长蛋白质形态。同样地,将β-连环蛋白的IntFOLD2模型与野生型β-连环蛋白的原始β-连环蛋白晶体结构叠合,以显示Thr653Lys突变残基。从所有可能旋转异构体中选取了最能体现Tyr654-p与Thr653Lys空间位阻冲突的侧链取向,该过程使用PyMOL的突变生成工具完成。
¶ 磁共振成像
对甲醛固定的小鼠脑标本进行了7特斯拉高分辨率T2加权和扩散加权磁共振成像,扫描参数如下:三维快速自旋回波( , ms,回波间隔 ,体素尺寸 );扩散加权图像(81个梯度方向, , , ,体素尺寸 , , )。实验采用雄性Bfc/+( 只)与对照野生型同窝仔 ; 只)。通过基于配准的方法计算解剖标记体积,使用2个平均参考模板( 组与对照组各一)。每个模板均经人工标注解剖区域,采用对称微分同胚配准算法获取个体特定解剖区域的体积(参考文献38)。扩散张量成像纤维追踪流程如下:首先对DWI图像进行重采样获得各向同性体素( ),手动去除非脑组织;随后使用FMRIB软件库对原始扩散数据拟合张量模型,生成分数各向异性图像v5.0 (FSL)。采用连续追踪算法的确定性纤维分配方法计算轴突纤维投射的估计值。纤维追踪终止标准包括各向异性阈值(低于0.15)和最大刚度条件,当连续步骤中的扩散方向差异超过 时即终止追踪。
¶ ASR与PPI
采用标准听觉惊吓设备(Med Associates)在Bfc/+( )和 ( 10)小鼠中测量了听觉惊吓反应(ASR)和前脉冲抑制(PPI)。惊吓反应测试环节包含5分钟适应期,随后给予5次110分贝白噪声脉冲。反应值以任意单位表示并取平均值。脉冲持续时间为10毫秒,平均试次间隔(ITI)为25秒(20-30秒)。PPI测试时,动物在接受110分贝主脉冲前会先接受60分贝前脉冲刺激。背景噪声设置为45分贝。前脉冲与主脉冲间隔为50毫秒。每种试验类型均以伪随机ITI重复呈现15次。实验在弱光环境下进行。通过前脉冲加惊吓试验类型测量PPI值,并以基础惊吓值的百分比表示(19)。
¶ 旋转杆
)和 )小鼠被要求在一根旋转棒上保持平衡。随后测量并评估它们从棒上跌落的潜伏期,以此作为其行为表现的衡量指标。在初始训练阶段(2分钟)中,所有小鼠先以恒定速度(每分钟4转[rpm])熟悉测试流程,随后启动从 加速至 的测试方案。我们将不同批次的 和 小鼠连续3天每天进行1次强化训练。每次训练包含6个连续阶段,每阶段持续1分钟:其中3个为从4 rpm加速至 的加速阶段,3个为保持 的匀速阶段。匀速阶段与加速阶段交替进行,所有小鼠均以匀速阶段开启当次训练。
¶ 超声波发声
当幼崽与巢穴、母鼠及同窝伙伴隔离时,会发出超声波叫声(39)。研究人员轻柔处理6日龄幼鼠(各基因型 ),每次从笼中单独取出一只放入隔离笼,该隔离笼内装有对超声波敏感的麦克风。该麦克风通过Avisoft UltraSoundGate 116 USB音频设备连接至个人电脑。监测在室温环境下进行,超声波记录时长为5分钟。
¶ WM
在本测试中, ( )与 ( )小鼠通过使用战略布置的视觉线索,被训练在充满不透明水体的水槽中定位水下或可见平台。实验开始前一天,将小鼠置于平台上进行1分钟的方向适应训练。随后在第1至4天,将小鼠放入水中并进行4次为一组的测试,组间间隔10至15分钟。可见平台被安置于略高于水面的位置,在规定时间内找到平台的小鼠需在平台上停留20秒。未能在规定时间内定位平台的小鼠则由人工引导至平台。每日测试结束时撤除平台,将小鼠置于水槽中观察1分钟(探查测试)。第5至8天期间,所有小鼠按上述方案进行每日训练,但此时平台被隐藏于水面之下。第9天变更平台位置后重复训练。通过视频追踪系统(Ethovision;Noldus)持续监测小鼠的自主活动情况。
¶ 恐惧条件反射
采用恐惧条件反射方案测试小鼠( 每组8只)的情境记忆能力。所有小鼠均通过多条件系统(TSE)进行测试。实验箱尺寸为 毫米(宽 深 高),室内光照强度维持在80勒克斯左右。经过120秒适应期后,小鼠接受28秒作为条件刺激的情境暴露,随后进行3次电击(2秒,0.5毫安)作为非条件刺激,每次刺激间隔90秒。记忆测试分别在24小时后(使小鼠重新暴露于相同情境5分钟)和48小时后(使小鼠重新暴露于线索刺激)进行。僵直行为定义为除呼吸外无任何动作,僵直时间的平均值以测试时长为基准按百分比表示。
¶ 时机学习
我们对小鼠进行了一系列认知计时测试,以评估其记忆奖励事件的能力及在时间维度上定位该事件的能力。本实验采用经典与新型(居家笼环境)操作式条件反射系统(TSE Systems),该系统基于动物的鼻触行为进行监测。
¶ 实验1:笼外测试与峰值程序任务Bfc/+()与()
小鼠按既往文献方法(40),连续8个工作日每天在经典操作条件反射箱中进行1小时训练。中央孔LED灯亮起伴随60分贝背景白噪音提示单次试验开始。当小鼠鼻触控制站时,中央孔LED与白噪音关闭,作为投食站的侧边目标孔则亮起20秒(图6A)。在最初10秒过后,安装在侧边孔的投食站会提供20毫克食物颗粒奖励。单次训练内的试验间隔为40至50秒。除奖励性试验外,还设置了一系列无奖励的探测试验。这些探测试验随机出现在各次训练中,平均每5次试验出现1次。
¶ 实验2:家笼自动化测试与切换任务
将6只Bfc/+和6只 小鼠饲养在家庭笼中(配备TSE新型三食槽操作墙),按先前描述(23)执行转换任务。简言之,所有试验均由动物通过鼻触家庭笼内1个食槽(位于操作墙中央)自行启动。奖励通过两侧食槽发放:1个食槽始终与短光信号关联,另1个始终与长光信号关联。仅当动物在特定信号后首次鼻触发生在正确位置时才能获得奖励。我们将所有小鼠训练至稳定表现状态;所有分析均保持短长信号1:2的比例(分别为3秒与6秒)。测试包含 无奖励(探针)试验以独立评估计时不确定性。错误次数、转换延迟、时间准确性和时间精度等指标描述了本测试中的计时认知内表型(如文献23、24所述)。
¶ 原代神经元培养
原代神经元培养在IIT和MRC同时进行。针对Bfc/+与WT神经元的研究,我们从E14.5期杂交背景 )母鼠胚胎中分离海马体。组织采集后置于冰上神经基础培养基(Invitrogen),经 胰蛋白酶( )消化20分钟,后续多次冲洗。铺板前通过轻柔地上下吹打重悬细胞。将细胞接种于包被多聚赖氨酸(Sigma-Aldrich)和层粘连蛋白(Invitrogen)的玻片上,置于24孔板内,使用神经生长培养基(含 -谷氨酰胺与 青霉素的神经基础培养基)进行培养。
青霉素/链霉素和 B-27补充剂,在标准细胞培养箱中。DiI (1,1′-二硬脂酰- -四甲基吲哚羰花青高氯酸盐;Invitrogen) 用于标记培养的神经元,样品随后用含有 DAPI 的 ProLong (Invitrogen) 封片介质包埋。染色的原代神经元使用 LSM 710 共聚焦显微镜 (Zeiss) 和 倍物镜(Plan-Apochromat 1.4) 进行观察。树突 每个基因型6个) 的长度和数量,针对 和 siRNA 培养物,使用相关软件 (ZEN 2010,版本 6.0.0.320) 进行分析。
¶ siRNA转染与qPCR表达检测
在小干扰RNA实验中,神经元取自胚胎期17.5天的C57BL/6J小鼠。培养1天的皮质神经元使用Lipofectamine RNAiMAX转染试剂(Invitrogen),按照制造商方案分别转染Silencer Select预设计siRNA靶向β-连环蛋白(siRNA编号:s63417)或对照siRNA(均购自Invitrogen)。在培养第7天检测细胞β-连环蛋白表达或神经突延伸情况。RNA提取、逆转录和定量RT-PCR操作参照先前文献(41)进行。采用GeNorm算法(42)的多内参基因法对基因表达数据进行标准化。 -连环蛋白(Ctnnb1)表达数据以Ppia和Gapdh参考基因进行归一化。引物序列如下:GAPDH(正向): -GAACATCATCCCTGCATCCA- ;GAPDH(反向): -CCAGTGAGCTTCCCGTTCA- ;PPIA(正向): -CACTGTCGCTTTTCGCCGCTTG- ;PPIA(反向): -TTTCTGCTGTCTTTGGAACTTTGTCTGC- ;Ctnnb1(正向): -CAATGGCTTGGAAATGAGACT- ;Ctnnb1(反向): -CCGTATCCACCAGAGTGAA-3′。
¶ 高密度MEA
高密度微电极阵列(MEA)平台(3Brain GmbH公司)已在先前论文(43, 44)中描述。简言之,这些MEA是单片CMOS器件,能够以 电极的采样频率同时记录来自4,096个电极的细胞外电生理信号。电极呈正方形(边长 ),以 阵列排列(间距 ,密度580电极 )。这些芯片能以空前的时空分辨率表征稀疏培养物的网络活动。取小鼠胚胎E14.5阶段的海马原代神经元进行分离,培养于APS-MEA芯片上。芯片经 乙醇消毒20分钟并包被聚乙亚胺(PEI)层。每个胚胎单独解剖,将 l细胞悬液滴加至各芯片记录区域(约200细胞 )。细胞贴壁后,芯片储液槽注入 Neurobasal培养基(Invitrogen公司),置于培养箱中直至记录。体外培养14天后,在基础状态及添加 BIC后分别进行10分钟网络活动记录。
在分析( , ; )过程中,我们采用精确时序尖峰检测(PTSD)算法进行峰值检测(45)并辅以定制Python脚本。所有对脉冲序列的后续分析均通过Python定制脚本实现。具体而言,通过单通道脉冲序列爆发检测器(46)识别爆发活动,并通过仅基于活跃通道(即发放率> )计算的MFR(平均发放率)、MBR(平均爆发率)和MBD(平均爆发时长)来表征全网络活动特性。功能连接性分析通过电极间脉冲事件互相关计算完成,并选取相关性最强的100至200个连接链路进行重点考察。
¶ 电子显微镜
和 )小鼠经 乌拉坦溶液(0.1毫升/10克)深度麻醉后,经心脏灌注冰生理盐水3至4分钟,随后灌注含 多聚甲醛和 戊二醛的0.1 M二甲胂酸钠缓冲液 。取脑组织后随后用振动切片机进行冠状切片( ),将感兴趣区域切成小块,并在 、 和0.1 M二甲胂酸钠中进行后固定,再用 醋酸铀的 乙醇溶液进行块染。最后将样品脱水并包埋于环氧树脂(Epon 812;TAAB公司)。超薄切片经 醋酸铀和佐藤氏柠檬酸铅染色后,使用JEOL JEM-1011显微镜在 加速电压下观察。图像通过ORIUS SC1000 CCD相机(Gatan公司)采集。采用ImageJ软件对突触面积、突触小泡数量和活性区长度进行量化分析。使用非配对学生t检验(GraphPad Prism 5.0)判定统计学差异。
¶ LTP实验
成年(12周龄)小鼠(每种基因型 只)经乌拉坦(Sigma-Aldrich公司)麻醉后断头处死。快速取出脑组织,用振动切片机(Leica公司VT 1000M型)在冰浴切割人工脑脊液(ACSF)中切片,该溶液成分为: 蔗糖、 、 葡萄糖、 、 、1 ,pH值7.3,并饱和充以 和 。将冠状海马切片(厚度 )在室温下于记录溶液中恢复至少2小时,该记录溶液成分为: 、 、 葡萄糖、 KCl、 、 和 ,pH值7.3,并饱和充以 和 。 电生理实验在浸没式记录槽中进行。使用充填记录用ACSF的玻璃微电极(电阻1-2兆欧)在CA1区辐射层记录场兴奋性突触后电位(fEPSPs)。通过同心双极钨丝电极(FHC公司)对谢弗侧支通路施加电流刺激脉冲,该刺激电极与记录电极间距至少300微米。以 频率监测基础突触传递至少10分钟(刺激强度设置为产生最大fEPSP斜率的刺激值的 )。长时程增强(LTP)通过θ串刺激(θ节律串刺激,4个串刺激,串间隔 ;每个串刺激包含4个 的脉冲串,脉冲串间隔 ;脉冲串时长30毫秒)或强直刺激 持续1秒)诱导。� 数据采集使用MultiClamp 700B放大器和pClamp 10软件(Molecular Devices公司)。fEPSPs分析通过Python自定义程序完成。配对脉冲比率计算为第二次刺激的fEPSP斜率与第一次刺激的fEPSP斜率之比。LTP在刺激给予后45分钟的时间窗内计算,时长为15分钟。统计分析使用GraphPad Prism软件,采用双因素方差分析结合Bonferroni事后检验及Student t检验计算统计学显著性。
¶ 统计数据
每次实验数据均采用适当的统计工具进行分析。采用双因素方差分析及事后比较检验,同时使用GraphPad Prism统计软件进行非配对学生t检验(双尾;置信区间 )。数据以均值 标准误表示。所有结果均通过在文中汇报各数据集所使用的相关统计量进行描述, <小于0.05视为具有统计学意义。
¶ 研究批准
所有动物实验均在英国根据医学研究理事会Responsibility in the Useof Animals for Medical Research(1993年7月)发布的指导原则及内政部项目许可证编号30/2198进行,在意大利则根据项目许可证106/2009–B开展。所有实验均符合国际动物伦理使用准则。针对人体研究,在纳入研究前均已获得所有参与者法定监护人的书面知情同意。本研究已获得当地伦理委员会批准(NL 13636.091.07)。
¶ 致谢
我们感谢J. Assad、T. Fellin、F. Benfenati、F. Szele、A. Greenfield和S. Brown对稿件进行的审阅。感谢A. Galbusera负责动物准备和磁共振图像采集。我们衷心感谢所有患者及其家属参与这些研究,并感谢Willemijn Wissink-Lindhout、Saskia van der Velde-Visser、Gaby van de Ven-Schobers和Marga Schepens提供的技术支持。此项工作获得了英国医学研究理事会(授予P.M. Nolan)、意大利技术研究院(授予V. Tucci)的资助,以及“独立自强”联盟(授予T. Kleefstra与M.H. Willemsen)、荷兰健康研究与发展组织(ZonMw)(编号907-00-365授予T. Kleefstra)、欧盟第七框架计划(Gencodys)的经费支持。
HEALTH-F4-2010-241995 授予 A. Vulto-vab Silfhout、Z. Iqbal与 T. Kleefstra),以及巴基斯坦伊斯兰堡高等教育委员会(授予 Z. Iqbal)。
通讯地址:Valter Tucci,意大利技术研究院神经科学与脑技术研究所,Via Morego 30号,热那亚 16163,意大利。电话:3901071781;传真:3901071781230;电子邮件:valter.tucci@iit.it。或联系:Patrick M. Nolan,英国医学研究理事会哈维尔研究所,哈维尔科学与创新园区,牛津郡 OX11 0RD,英国。电话:441235841200;传真:441235841200;电子邮件:p.nolan@har.mrc.ac.uk。
- 贾米森 C、夏尔马 M、亨德森 BR. Wnt 信号从膜到核:β-catenin 陷入循环. Int J Biochem Cell Biol, 2012, 44(6):847-850.
- 邢宇等. 全长β-连环蛋白的晶体结构. Structure, 2008, 16(3):478-487.
- 格里戈良 T、温德 P、克劳斯 A、伯奇迈尔 W. 解读经典Wnt信号在发育和疾病中的功能:小鼠中β-catenin的条件性功能丧失和功能获得突变. Genes Dev, 2008, 22(17):2308-2341.
- Salinas PC, Price SR. 突触发育中的钙黏蛋白和连环蛋白. Curr Opin Neurobiol, 2005, 15(1):73-80.
- Huber AH, Weis WI. β-连环蛋白/E-钙黏蛋白复合物的结构及β-连环蛋白多样配体识别的分子基础. Cell, 2001, 105(3):391-402.
- 穆勒 T、乔伊达斯 A、赖希曼 E、乌尔里希 A. 磷酸化和β-连环蛋白的游离池受酪氨酸激酶和酪氨酸磷酸酶在上皮细胞迁移过程中调节. J Biol Chem, 1999, 274(15):10173-10183.
- 帕特雷 P 等. PTP1B 调控由细胞间和细胞-基质粘附分子介导的神经突延伸. J Neurosci Res, 2001, 63(2):143-150.
- Murase S, Mosser E, Schuman EM. 去极化驱动β-连环蛋白进入神经元树突棘,促进突触结构和功能的改变. Neuron, 2002, 35(1):91-105.
- 坎德尔 ER. 《记忆存储的分子生物学:基因与突触的对话》. Science, 2001, 294(5544):1030-1038.
- Maguschak KA, Ressler KJ. β-连环蛋白是记忆巩固所必需的分子. Nat Neurosci, 2008, 11(11):1319-1326.
- O’Roak BJ 等. 多重靶向测序识别自闭症谱系障碍中复发性突变基因. Science, 2012, 338(6114):1619-1622.
- O’Roak BJ 等. 散发性自闭症外显子组揭示了一个由de novo突变组成的高度互连蛋白质网络. Nature, 2012, 485(7397):246-250.
- de Ligt J 等. 重症智力障碍患者的诊断性外显子组测序. N Engl J Med, 2012, 367(20):1921-1929.
- Nolan PM 等. 一项系统的、全基因组范围的、表型驱动的小鼠基因功能研究诱变计划. Nat Genet, 2000, 25(4):440-443.
- Fossat N 等. 小鼠胚胎头部形成对经典WNT信号通路活动适当水平的严格要求. Development, 2011, 138(4):667-676.
- Lilien J, Balsamo J. 酪氨酸磷酸化/去磷酸化对β-连环蛋白介导的钙黏蛋白粘附的调控作用. Curr Opin Cell Biol, 2005, 17(5):459-465.
- 徐 W、基梅尔曼 D. 机制性见解:来自β-连环蛋白及其结合蛋白的结构研究. J Cell Sci, 2007, 120(19):3337-3344.
- Chenn A, Walsh CA. 通过控制神经前体细胞周期退出调节大脑皮层尺寸. Science, 2002, 297(5580):365-369.
- Mandillo S 等. 小鼠行为表型分析中的可靠性、鲁棒性与可重复性:一项跨实验室研究. Physiol Genomics, 2008, 34(3):243-255.
- Morris R. 用于研究大鼠空间学习的水迷宫程序开发. J Neurosci Methods, 1984, 11(1):47-60.
- Johansen JP 等. 恐惧学习与记忆的分子机制. Cell, 2011, 147(3):509-524.
- Gallistel CR 等. 小鼠计时行为变异性与系统误差的来源. J Exp Psychol Anim Behav Process, 2004, 30(1):3-16.
- Lassi G 等. Gnas印记缺失对小鼠REM/NREM睡眠及认知的差异性影响. PLoS Genet, 2012, 8(5):e1002706.
- Balci F 等. 人类与小鼠的风险评估. Proc Natl Acad Sci U S A, 2009, 106(7):2459-2463.
- O’Roak BJ 等. 多重靶向测序鉴定自闭症谱系障碍中的复发突变基因. Science, 2012, 338(6114):1619-1622.
- Cao F 等. 自闭症患者额叶皮质中星形胶质细胞及Wnt/β-catenin信号通路的改变. J Neuroinflammation, 2012, 9(1):223.
- Campos VE 等. β-catenin突变小鼠的癫痫易感性增加与皮质畸形. Biochem Biophys Res Commun, 2004, 320(2):606-614.
- Machon O 等. β-catenin在发育中皮质与海马神经上皮中的作用. Neuroscience, 2003, 122(1):129-143.
- Schüller U, Rowitch DH. 小脑形态发生需要β-catenin功能. Brain Res, 2007, 1140:161-169.
- Huelsken J 等. 小鼠前后轴形成对γ-catenin的需求. J Cell Biol, 2000, 148(3):567-578.
- van Veelen W 等. β-catenin酪氨酸654磷酸化增强Wnt信号与肠道肿瘤发生. Gut, 2011, 60(9):1204-1212.
- Sharma M 等. 特异性犰狳重复序列通过直接结合核孔蛋白Nup62、Nup153与RanBP2/Nup358促进活细胞中β-catenin核转运. J Biol Chem, 2012, 287(2):819-831.
- Piedra J 等. 酪氨酸磷酸化对β-catenin结构与活性的调控. J Biol Chem, 2001, 276(23):20436-20443.
- Yu X, Malenka RC. β-连环蛋白对树突形态发生至关重要. Nat Neurosci, 2003, 6(11):1169-1177.
- Buenavista MT, Roche DB, McGuffin LJ. 基于单模板模型质量评估的多模板引导优化三维蛋白质模型. Bioinformatics, 2012, 28(14):1851-1857.
- Roche DB, Buenavista MT, Tetchner SJ, McGuffin LJ. IntFOLD服务器:集成式蛋白质折叠识别、三维模型质量评估、内在无序预测、结构域预测及配体结合位点预测网络资源. Nucleic Acids Res, 2011, 39(网络服务器版):W171-W176.
- Esapa CT 等. 导致肌阵挛-肌张力障碍综合征的SGCE错义突变会损害ε-肌聚糖向质膜的运输:泛素化及torsinA的调控作用. Hum Mol Genet, 2007, 16(3):327-342.
- Avants BB, Epstein CL, Grossman M, Gee JC. 基于互相关的对称微分同胚图像配准:评估老年性与神经退行性脑部自动标记. Med Image Anal, 2008, 12(1):26-41.
- Scattoni ML, Crawley J, Ricceri L. 超声波发声:神经发育障碍小鼠模型行为表型分析工具. Neurosci Biobehav Rev, 2009, 33(4):508-515.
- Tucci V, Hardy A, Nolan PM. C57BL/6J小鼠食物限制与饮水限制方案下的生理和行为参数比较. Behav Brain Res, 2006, 173(1):22-29.
- Giacomini C 等. 雪旺细胞与轴突缺陷共同导致Ebf2-/-小鼠运动周围神经病变. Neurobiol Dis, 2011, 42(1):73-84.
- Vandesompele J 等. 通过多内参基因几何平均实现实时定量RT-PCR数据的精准标准化. Genome Biol, 2002, 3(7):RESEARCH0034.
- Berdondini L 等. 用于从单细胞到大规模神经元网络的高时空分辨率电生理记录的有源像素传感器阵列. Lab On A Chip, 2009, 9(18):2644-2651.
- Imfeld K 等. 用于细胞外电生理活动记录的大规模高分辨率数据采集系统. IEEE Trans Biomed Eng, 2008, 55(8):2064-2073.
- Maccione A, Gandolfo M, Massobrio P, Novellino A, Martinoia S, Chiappalone M. 细胞外记录神经元信号中峰值精准识别的新算法. J Neurosci Methods, 2009, 177(1):241-249.
- Chiappalone M, Novellino A, Vajda I, Vato A, Martinoia S, van Pelt J. 用于分析皮层神经元网络时空模式的爆发检测算法. Neurocomputing, 2005, 65-66:653-662.