¶ 共济失调毛细血管扩张症患者来源的神经元及脑类器官模型揭示线粒体功能障碍与氧化应激
¶ Ataxia Telangiectasia patient-derived neuronal and brain organoid models reveal mitochondrial dysfunction and oxidative stress
汉娜·C·利森a*、胡里奥·阿瓜多a、塞西莉亚·戈麦斯-因克兰、哈曼·考尔·查格a、阿特法·塔赫里安·法德a、佐伊·亨特a、马丁·F拉文b、艾伦·麦凯-西姆、厄恩斯特·J沃尔维唐a*
原文下载:https://fb.biokingdom.top/f/e9b35c441664471a8e10/
¶ 文章信息
关键词:共济失调毛细血管扩张症、脑类器官、线粒体、神经退行性变、细胞衰老、氧化应激
¶ 摘要
共济失调毛细血管扩张症(AT)是一种由ATM基因突变引起的罕见疾病,其导致的进行性神经退行性变机制至今尚未明确。除在核DNA修复中发挥核心作用外,ATM蛋白还在细胞核外调控新陈代谢、氧化还原稳态及线粒体功能。然而,在AT相关人类神经元模型中,关于ATM缺失如何及何时影响这些参数的系统性研究尚属空白。为此,我们利用AT患者iPSC来源的皮质神经元与脑类器官及经基因校正的同基因对照组,揭示了线粒体功能障碍、氧化应激及细胞衰老水平随发育成熟度变化的规律。转录组分析发现线粒体功能与维持相关调控网络异常,包括PARP/SIRT信号轴改变以及关键线粒体自噬与线粒体分裂-融合过程失调。我们进一步证明抗氧化剂可降低AT神经元培养物中的活性氧并恢复神经元分支,同时改善AT脑类器官中受损的神经元活动。本研究得出结论:进行性线粒体功能障碍与异常活性氧产生是AT神经退行性变的重要诱因,且与ATM在线粒体稳态调节中的作用密切相关。
¶ 1.引言
共济失调毛细血管扩张症(AT)是一种罕见的常染色体隐性遗传病,由共济失调毛细血管扩张突变基因(ATM)的变异引起。该基因编码的3 丝氨酸/苏氨酸激酶参与DNA损伤应答和抗氧化防御通路。临床症状包括免疫缺陷、癌症风险升高、早衰以及由小脑进行性神经退行性病变引发的共济失调(Stracker等,2013;Shiloh与Lederman,2017)。健康个体中,ATM激酶会因DNA双链断裂激活,作为复杂信号轴的核心调控因子协调DNA修复、细胞周期阻滞或凋亡(Bakkenist与Kastan,2003;Shiloh与Ziv,2013)。尽管神经元不进行DNA复制,但DNA双链断裂仍可能由生理性神经元活动(Madabhushi等人,2015),以及源于功能失调的线粒体和活性氧[ROS;(Vizioli等人,2020)]产生的线粒体至细胞核逆行信号级联。然而,有缺陷的ATM如何导致AT患者小脑和海马体中有丝分裂后细胞的退化仍不明确,对此的争论持续不断:这究竟仅归因于有缺陷的DNA损伤反应(Shiloh,2020),抑或还有其他机制在起作用,例如异常的细胞周期重启(Rimkus等人,2008)、氧化损伤(Ditch和Paull,2012)或突触功能受损(Li等人,2009)。
ATM在线粒体稳态和氧化应激调节中的重要作用得以揭示,源于在A 小鼠小脑中发现活性氧水平升高的研究结果(Kamsler等人,2001;Quick与Dugan,2001;陈等人,2003)。升高的氧化应激与在AT患者的淋巴细胞、永生化淋巴母细胞(Reichenbach etal.,2002;Ambrose etal.,2007)以及成纤维细胞中均检测到线粒体功能紊乱,其中成纤维细胞还表现出线粒体自噬水平下降(Valentin-Vega etal.,2012)。这一发现尤其值得关注,因为线粒体在神经元功能中扮演重要角色,而小脑对线粒体损伤具有特殊敏感性(Lax etal.,2012;Lopriore et al.,2022)一一任何线粒体稳态的偏离都可能对神经元完整性和功能产生负面影响(Kann and Kovacs,2007)。ATM蛋白定位于线粒体,可在无需DNA损伤应答通路激活的情况下,通过线粒体依赖性方式被氧化应激直接激活(Valentin-Vega etal.,2012;Guo etal.,2010;Morita et al.,2014),这表明ATM还能作为活性氧传感器介导抗氧化反应。氧化的ATM会形成二硫键交联二聚体(Guo etal.,2010),当被氧化应激激活时,其作用靶点蛋白超过2500种(Kozlov etal.,2016),而响应DNA损伤的靶点蛋白仅700种(Matsuoka et al.,2007)。
尽管如此,目前仅有有限的研究致力于探讨非转化人类神经元模型中ATM蛋白的功能(Corti等人,2019;Lee等人,2013),且均未深入探索线粒体功能障碍。虽然ATM缺陷小鼠能重现部分共济失调毛细血管扩张症(AT)的细胞缺陷,但其他AT相关缺陷(如神经元退行性变)在ATM-/小鼠模型中并不明显(Lavin,2013)。因此,利用AT患者诱导多能干细胞(iPSC)分化的神经元模型,为在相关非转化人类神经系统中研究ATM在线粒体功能障碍中的作用提供了契机。本研究通过量化不同成熟阶段AT患者神经模型(包括原代组织、iPSC、iPSC源性神经祖细胞、二维神经元培养及脑类器官)的线粒体含量、膜电位和氧化应激水平,系统绘制了线粒体功能障碍的特性与时序调控图谱。我们探索了与线粒体功能相关的神经元活动及衰老表型,揭示了调控线粒体过程的关键基因调控网络,并指出若干可能促成AT神经元退行性变的潜在机制。
¶ 2.方法
除非另有说明,所有试剂均购自Thermo Fisher Scientific。
¶ 2.1.人嗅觉上皮细胞培养
共济失调毛细血管扩张症(AT)患者于澳大利亚布里斯班昆士兰大学临床研究中心的AT专科门诊确诊。依照先前所述方法(Stewart等,2013),在获得知情同意后采集了五名AT患者和六名年龄匹配的健康对照者的嗅黏膜活检组织,并将获得的嗅上皮细胞置于DMEM/F12培养基中培养,该培养基添加了 胎牛血清、1:100GlutaMAX、1:100非必需氨基酸及 青霉素-链霉素。当细胞融合度达到 时,约每5天进行一次传代培养。
¶ 2.2.诱导多能干细胞培养与神经元分化
本研究使用了四种iPSC系:源自ONS细胞的AT与对照iPSC(Leeson等,2021a;Leeson等,2021b),分别称为“AT”和“对照”;以及AT32患者及其同基因型(基因校正)iPSC配对(Nayler等,2017;Ovchinnikov等,2020;Nayler等,2012)。ATM基因突变描述见附图3。iPSC在经hESC认证基质胶(康宁)包被的培养板上,使用mTeSRPlus(STEMCELLTechnologies)培养基进行培养。当细胞融合度达? 时,每5天左右使用EDTA进行团块传代。库存细胞使用 Synth-a-Freeze 以细胞团块形式冷冻保存,并在ROCK抑制剂Y-27632( ;Tocris)存在条件下进行解冻。
iPSC细胞系采用改良自Shi、Kirwan(Shi等,2012)的方案分化为神经元。简言之,将iPSC培养至 汇合度后,更换为3N神经分化培养基(DMEM/F12与Neurobasal培养基按1:1混合,添加1:200 N2、1:100 B-27、1:100 GlutaMAX、1:200 NEAA、 胰岛素、 -巯基乙醇及 PenStrep),并加入SB431542( ;美天旎)和LDN-193189( ;Sigma)。每日更换含SB与LDN的3N培养基,持续10天,随后使用Dispase II(2.4单位 )消化细胞,按1:3比例重铺于基质胶包被培养板的3N培养基中。神经上皮细胞隔日换液,神经玫瑰花环结构出现时添加 FGF2(普洛麦格)。人工筛选含神经祖细胞 (NPC)的玫瑰花环结构,后续传代均在 汇合度时使用Accutase进行。NPC可培养为2D单层(铺于基质胶包被板)或3D神经球(低吸附培养板)。为诱导神经元成熟,将NPC(P2起)以50,000细胞 密度接种于PLO-层粘连蛋白包被板或盖玻片,并在3N神经分化培养基中添加抗坏血酸(200nM;Sigma)、二丁酰-cAMP( ;Sigma)、BDNF( 普洛麦格)、GDNF( ;普洛麦格)及DAPT( ; Sigma)。神经元培养2至10周,每周3次更换 培养基。部分实验中对神经元培养物添加ATM抑制剂KU-55933或KU-60019(Sigma,各 ,每周3次随换液添加)、抗氧化剂N-乙酰半胱氨酸(NAC; 每日)或回补剂庚酸(C7; ,每周3次随换液添加)。所有处理在4周评估前持续10-12天。
¶ 2.3.脑类器官的生成
利用人类AT32突变型和基因校正型iPSC生成脑类器官。使用Accutase解离iPSC,以每孔15,000个细胞的密度接种于96孔低吸附U底板中,培养基为含 ROCK抑制剂的mTeSRPlus。 离心5分钟促使细胞聚集。球体隔夜形成后,连续5天每日更换KSR培养基[DMEM/F12、 Knock-out血清替代物(KSR)、1:100 GlutaMAX、1:200 MEM-NEAA、Dorsomorphin ( ;StemMACS)和A-83-01( ;Lonza)]。第6天更换为诱导培养基(含DMEM/F12、1:100 N2、1:100 GlutaMAX、1:200 MEM-NEAA、 肝素(Sigma)、 CHIR 99021(Lonza)和 SB-431542 (Sigma))。第11天将球体包埋于基质胶( 液滴, 凝胶化20分钟)并转移至含诱导培养基的低吸附24孔板。从第16天起更换为类器官培养基(DMEM/F12与Neurobasal培养基1:1混合,添加1:100N2补充剂、1:50B27补充剂、1:100 GlutaMAX、1:200 MEM-NEAA、 青霉素-链霉素、 2-巯基乙醇和 胰岛素),每周换液三次直至包埋后100天取材。
¶ 2.4. 免疫化学
所有样品均使用 多聚甲醛固定。细胞和类器官切片经 Triton-X100渗透化处理后,用 BSA封闭1小时。一抗于 条件下孵育过夜,二抗在室温下孵育1小时。所用抗体均列于补充材料表S1。封片前采用DAPI进行细胞核复染。图像采集使用奥林巴斯BX61或蔡司AxioScan系统,并通过FIJI(ImageJ)或Cell Profiler软件对获取图像进行分析。
¶ 2.5.线粒体功能检测与Operetta高内涵成像系统
采用MitoTrackerTMDeepRedFM活细胞染色剂( ,孵育30-40分钟)测定总线粒体含量。使用四甲基罗丹明乙酯(TMRE;艾博抗公司, ,20分钟)检测线粒体膜电位,并通过MitoTrackerTMRed CM-H 评估活性氧产生量—一这是一种经还原处理的非荧光罗胺染料,在被活性氧物种氧化后发荧光,随后被线粒体捕获( ,30-45分钟)。孵育完成后,对细胞进行清洗并用Hoechst33342进行复染,再次清洗后置于不含酚红的培养基中进行成像。使用OperettaCLS高内涵分析系统(珀金埃尔默公司)采集图像。每个实验至少进行3次生物学重复。
¶ 2.6.基于Harmony的无偏图像定量分析
采用Harmony软件对OperettaCLS高内涵分析系统捕获的线粒体染色进行无偏倚自动定量。分析流程基于预定义的Harmony算法进行修改,并对所有图像应用高级平场校正。细胞核区域通过Hoechst或DAPI染色确定,并根据直径、分割灵敏度及通用阈值需求进行调整。通过阈值优化识别细胞核周围的细胞体区域,胞质区域通过从细胞体区域扣除细胞核区域计算获得。边界物体及不规则或死亡细胞均被排除。计算选定物体每像素单元的荧光强度。对于神经元培养体系,设定阈值将胞质ROI限制在核周区域,采用点状检测算法识别神经元突起沿线的线粒体斑点。对于ONS、iPSC和NPCs,通常每个96孔板孔捕获25-40个视野,每个视野定量约200-500个细胞。对于神经元培养体系,24孔板每孔通常采集50-80个视野,捕获200,000至400,000个细胞,Find Spots算法在每个视野中通常可识别约300,000至600,000个斑点。视野结果取平均值后以整孔数据形式输出(各孔以均值 标准差表示)。所有统计学分析与结果解读均基于整孔数据开展。
¶ 2.7.衰老相关β-半乳糖苷酶检测(SA-β-Gal)
神经元二维培养物经PBS清洗后,用 多聚甲醛固定10分钟,再次清洗,随后在 (无二氧化碳条件下)与新鲜配制的SA -Gal染色液(p )共同孵育。染色液组成:铁氰化钾 、亚铁氰化钾 、磷酸二氢钠 、磷酸氢二钠 、氯化钠 、二氯化镁 以及5-溴-4-氯-3-吲哚 -D-半乳糖苷 。染色反应在2-4小时内显现,并于12-16小时达到峰值。
¶ 2.8.Western印迹法
细胞用PBS冲洗后,使用含有蛋白酶和磷酸酶抑制剂(罗氏)的RIPA缓冲液 ( Tris , 氯化钠、 Triton X-100、 脱氧胆酸钠、 SDS)进行裂解。针对总ATM检测的样本制备时,取 蛋白质与DTT( 及 Laemmli SDS上样染料混合, 加热10分钟。而采用OXPHOS抗体混合液检测的样本因复合体IV的MTCO1对热高度敏感而未进行加热处理。裂解液在变性条件下通过SDS-PAGE进行分离,并转印至硝酸纤维素膜。对于OXPHOS抗体混合液检测,使用高pH值CAPS转印缓冲液( CAPS[3-(环已氨基)-1-丙磺酸]、 甲醇、 )将蛋白转印至PVDF膜。转印后的膜经过封闭处理。在含有 脱脂奶粉的TBS-Tween中孵育1小时。一抗混合物在封闭缓冲液中稀释,并在4 下孵育过夜。膜被洗涤,并用HRP标记的二抗在室温下探测一小时。使用Clarity ECL(Bio-Rad)或Femto最大灵敏度底物检测OXPHOS混合物的交叉反应性。捕获的图像使用Image Lab 4.1(Bio-Rad)软件进行分析。蛋白质表达水平归一化至上样对照(β-肌动蛋白或 微管蛋白)。未裁剪的印迹图提供在补充数据中。
¶ 2.9.qPCR
使用RNeasyMiniKit(Qiagen)按照制造商说明从培养细胞和脑类器官中提取总RNA。取1μg总RNA采用iScriptcDNA合成试剂盒(Bio-Rad)进行反转录。qPCR使用PowerUp SYBR Green预混液(Applied Biosystems)在Bio-RadCFX96Touch实时PCR检测系统上进行。每个反应设置2个技术重复及3个生物学重复。以ETFA或GAPDH作为内参基因。引物序列列于附表S2。
¶ 2.10. RNA测序
对AT32突变型和基因校正的2周龄神经元(各3个生物学重复)及100天龄脑类器官(各4个类器官)进行RNA测序。在由诺禾致源(香港)公司进行测序前,通过2100 BioanalyzerRNA6000 Pico Chip芯片试剂盒(安捷伦)采用RNA完整值(RIN)确认RNA完整性。使用NEBNext?UltraTMRNA文库构建试剂盒(美国NEB)制备总RNA文库,并在Illumina NovaSeq150 bp双末端测序平台上进行测序。分析前采用Fastp对原始序列进行质量检测以确认数据完整性。使用HISAT2软件将双末端清洁读数映射至人类基因组组装hg38,通过Featurecounts对每个基因(包括已知基因和新发现基因)的映射读数进行计数,并依据基因长度及映射至该基因的读数计数计算各基因的RPKM值。采用DESeq2R软件包进行组间差异表达分析,所得P值通过Benjamini-Hochberg方法校正以控制错误发现率(FDR)。经DESeq2鉴定且校正后P(padj)值 的基因被判定为差异表达基因(DEG)。基于上调或下调的差异表达基因,使用注释、可视化与集成发现数据库 (Sherman等,2022)进行基因本体 (GO术语)富集分析和KEGG通路分析。脑类器官RNA测序数据已存入欧洲核苷酸档案库,主要登录代码为PRJEB72015。
¶ 2.11.多电极阵列
对成熟至少100天的类器官采用高分辨率(4096电极)BioCamX MEA平台(3Brain,瑞士)进行多电极阵列分析。类器官在记录前3天被转换至含NeuroCult SM1神经元补充剂(StemCell Technologies)的BrainPhys培养基,并固定于BioCamX芯片上,采样频率为 采集数据后,使用集成BrainWave5软件进行滤波处理,识别峰电位(活动强度为基线>8个标准差)及簇发放电( 个峰电位且最大间隔100毫秒)。通过BrainWave5计算峰电位频率(平均发放率;MFR)、振幅、持续时间和事件间期。未接触类器官或记录到MFR低于 个/秒的电极被排除分析。在不同类器官上进行至少3次独立记录(时长3分钟),并在第1分钟时间点施加谷氨酸( )。谷氨酸处理后的MFR均以处理前 (基础)水平进行标准化 -乙酰半胱氨酸(NAC; 或庚酸盐(C7; 处理在记录前应用了两周。BioCamX数据表示为 标准误。
¶ 2.12. 统计分析
除非另有说明,每次实验至少进行3次生物学重复(N指从iPSCs阶段开始的分化实验),并包含至少2次技术重复! 指平行培养孔或同一NPC培养物同步进行的分化实验)。统计学显著性通过t检验 (独立样本)或单因素方差分析判定,事后分析采用TukeyHSD法。当Levene方差齐性检验显示方差存在显著影响时,则采用更严格的Welch单因素方差分析,并使用Games-Howell事后检验以降低第一类错误发生概率。 数值均保留三位小数直接呈现。数据分析使用IBMSPSS Statistics 27或GraphPad Prism完成。
¶ 3.结果
¶ 3.1.来自AT患者的原代嗅觉上皮细胞未表现出线粒体功能障碍
为研究AT患者与对照组患者原代组织中线粒体功能的疾病特异性差异,我们首先利用从原发性嗅上皮黏膜获取的人嗅上皮细胞(ONS;附图1A-C)。这类细胞具备神经上皮细胞的某些特征(Stewart etal.,2013),并在包括AT在内的多种神经系统疾病中呈现疾病特异性差异(Stewart etal.,2013;Mackay-Sim,2012)。我们通过定量分析总线粒体含量与线粒体膜电位(附图1D、E),并采用MitoTrackerRedCM-H2Xros检测活性氧簇生成量(附图1F)。基于无偏分析流程(附图1G),我们发现对照组与AT组ONS细胞在上述参数中均无显著差异。蛋白质印迹实验进一步显示两组样本在单个氧化磷酸化复合物表达上未见差异(附图2),由此得出结论:与对照组相比,AT患者来源的ONS细胞在线粒体含量、线粒体呼吸链复合体表达、膜电位及线粒体活性氧生成方面均未发生改变。
¶ 3.2.线粒体功能紊乱在AT干细胞和神经前体细胞群体中出现
鉴于神经元退行性变是共济失调毛细血管扩张症(AT)中最具致残性的病理特征之一,且目前仍是认知最薄弱的环节,我们随后在相关神经元模型中研究了线粒体功能障碍。为此,我们将AT患者嗅鞘神经细胞(ONS)重编程为诱导多能干细胞(iPSCs),旨在研究患者来源神经元及脑类器官中的线粒体功能障碍。参照既往方法(Leeson等,2021a;Leeson等,2021b),将AT患者ONS3号细胞系与对照组ONS重编程为iPSCs。这些细胞系及我们先前构建的AT32患者来源iPSCs与其同基因(基因校正)配对株系(Nayler等,2017;Ovchinnikov等,2020;Nayler等,2012)均携带导致截短型ATM蛋白失活的突变[附图3;(Leeson等,2021a;Ovchinnikov等,2020)]。蛋白质印迹分析证实,AT患者ONS来源iPSCs及AT32突变iPSC克隆株系中ATM表达量降低,而AT32基因校正可使表达水平恢复至对照组ONS来源iPSCs相当的程度(图1A)。
随后,我们对ONS来源的AT细胞系与对照iPSC细胞系中的线粒体含量、膜电位和氧化应激水平进行了定量分析(附图3D)。结果显示,与对照组iPSCs相比,AT细胞的MitoTracker和TMRE荧光强度均出现下降(图1B-D)。通过MitoTrackerDeep Red染质的纹理分析发现,AT细胞线粒体呈现出结构改变的特征与对照iPSC系的线粒体相比,其结构和/或亚细胞网络存在差异。ATiPSCs中也观察到氧化应激增加(图1E),这与先前研究结果一致(Ovchinnikov等人,2020)。随后将AT、对照及基因校正的iPSC分化为神经祖细胞(NPCs)和神经元(Shi等人,2012),这些细胞表达水平相当的祖细胞和早期神经元标志物(附图4)。在ATNPCs中未检测到线粒体含量的显著差异(图1F、G),但与各自对照组相比,两个AT患者系的膜电位均显著降低(图1H、I)。此外,ATM缺陷的NPC培养物中氧化应激水平升高,ONS来源的AT突变NPCs和AT32突变NPCs的CM 平均荧光强度较对照组分别增加 和 (图1J、K)。
我们得出结论,与对照组相比,ATiPSC的线粒体数量更少、结构改变,且活性氧(ROS)生成量略有增加。此外,ATiPSC在向NPC分化过程中未表现出缺陷。与无关及同基因对照组相比,ATNPC虽未出现线粒体含量异常,但确实表现出线粒体膜电位降低和ROS生成增加,这与在未分化iPSC中观察到的现象相似。鉴于线粒体膜电位存在显著缺陷且ROS生成增加,我们推测这可能会对神经元成熟过程中的线粒体稳态产生下游影响。
¶ 3.3. AT患者iPSC衍生的神经元显示线粒体缺陷且定位异常
AT及其各自的对照/同基因校正神经元成熟了2-4周(图2A),到2周时 的细胞表达神经元标志物βIII微管蛋白和Map2(图2B)。AT突变神经元出现与神经元变性一致的起泡和碎裂现象,这在βIII微管蛋白染色后最为明显。我们分别测量了2周和4周龄神经元胞体(神经元细胞体)及神经突(神经突触)中线粒体染料的荧光强度(附图5A)。2周龄AT神经元表现出胞质MitoTracker强度降低而神经突强度升高(附图5B、C),表明线粒体正定位于神经元外周。线粒体膜电位与MitoTracker观察结果相关(附图5D、E)。与神经祖细胞培养组不同,2周龄神经元中AT32突变与同基因校正神经元的氧化应激水平未呈现显著差异(附图5F、G)。
成熟至第4周时,神经元培养物呈现出与较年轻的2周龄对照组不同的线粒体表型。ONS-iPSC来源的AT神经元表现出线粒体定位和/或运输功能受损,具体表现为胞质区域总线粒体染色减少(图2C、D)以及神经突中线粒体斑点数量增加 (补充图7A),而神经突斑点强度保持不变。我们推测这可能与线粒体更新及受损线粒体回收机制障碍有关,这与ATM蛋白作为线粒体自噬调节因子(Valentin-Vega等,2012;Ciroti等,2021;Yeo等,2021a;Sarkar等,2021)和线粒体分裂调节因子(Luo等,2023)的功能相符。相比之下,AT32突变神经元在保持稳定斑点数量的同时,其神经突内线粒体含量显著减少(图2C、E,补充图6A)。这同样提示线粒体结构排布与分裂/融合过程存在异常,不过两种情形可能并存并共同导致线粒体功能障碍的整体表型,不同细胞系间的差异很可能反映了患者特异性特点。TMRE检测持续显示4周龄神经元培养物存在线粒体膜电位紊乱(图2F-H),这与在NPCs和2周龄神经元中的观察结果相似,且在胞质线粒体荧光强度方面尤为明显(图2G)。
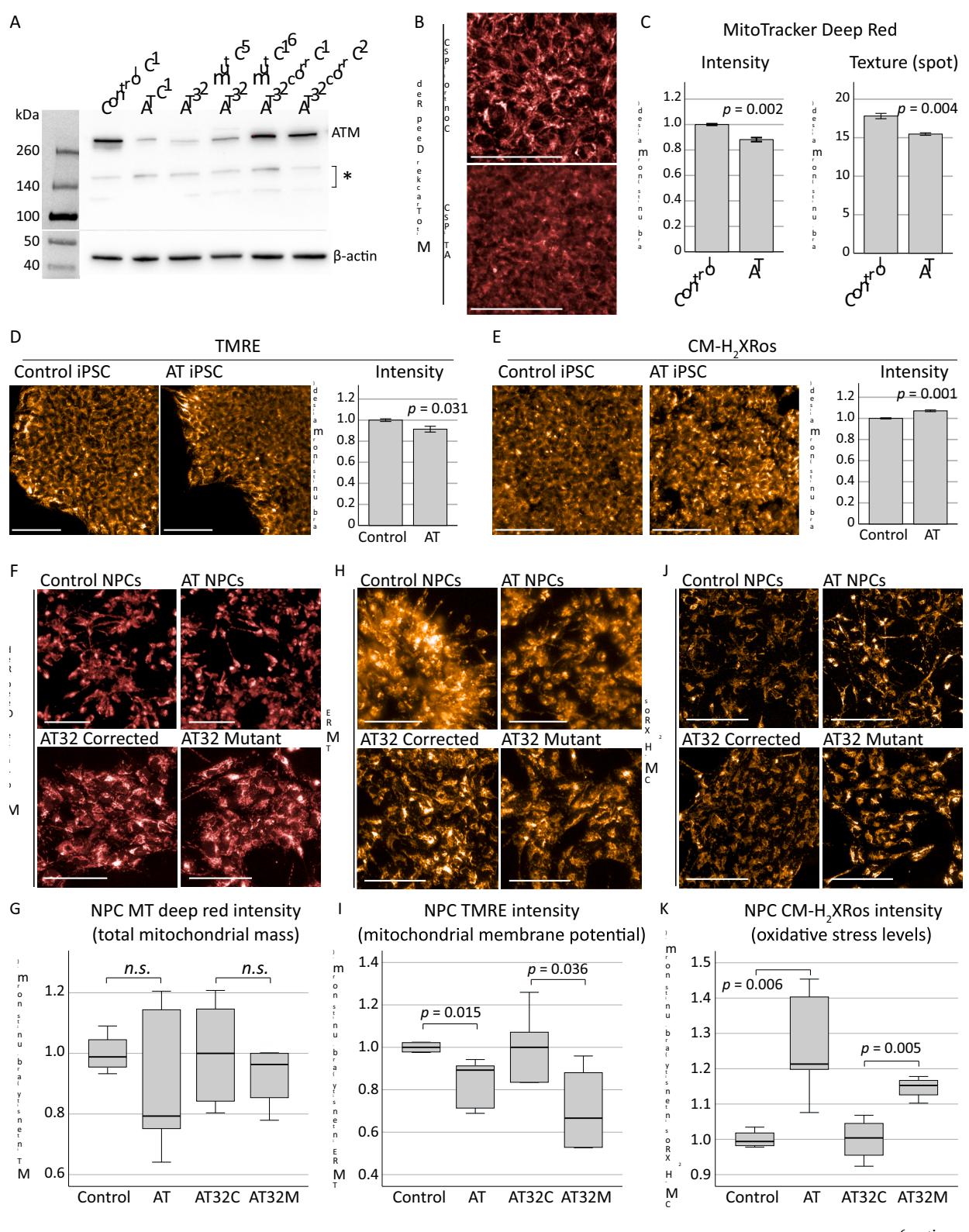
图1.对来自AT患者的iPSCs和NPCs进行的线粒体评估。
A)Westemblot结果证实,与对照组(对照克隆C1)相比,ATONS来源iPSC系(AT克隆1[C1])及两个AT32iPSC系克隆(突变克隆C5/C16,校正克隆CI/C2)中ATM蛋白水平降低。AT32系的基因校正使ATM表达恢复至对照组相当水平。星号标示非特异性条带,C代表克隆编号。B-E)通过MitoTracker Dep Red和TMRE评估AT与对照iPSC系的线粒体含量与功能,采用CM s检测氧化应激水平。MitoTracker Deep Red(B,C)显示线粒体含量显著降低 ,染色纹理分析表明ATiPSC的线粒体结构发生改变 ,该分析采用unbiased 算法量化各荧光斑点的鞍点/斑点/脊变量。TMRE(D)显示ATiPSC膜电位显著降低 , (E)检测表明ATiPSC氧化应激水平升高 。误差棒表示均值 标准误。F-K)将AT、对照及AT32 iPSC分化为NPC后评估线粒体参数。MitoTracker Deep Red(F-G)显示对照组与ATNPC线粒体含量无显著差异 ,AT32突变型与基因校正NPC间亦无差异( 。TMRE(H-I)表明AT与AT32突变系NPC膜电位均显著低于对照及AT32校正组(相应值: .036)。J-K) Xros检测显示两个AT系氧化应激水平均显著升高(AT组对照: ;AT32配对: 。柱状图为均值±标准误,箱形图为中位数 四分位距,须线表示极值。所有比例尺为 AT指ATONS来源iPSC系;Control指对照ONS来源iPSC系;AT32C指AT32基因校正iPSC系;AT32M指AT32突变iPSC系;C为克隆编号。 (关于图例色彩说明请参阅本文网络版)
有趣的是,当我们在培养4周的神经元中使用Mito-Tracker CN 评估氧化应激水平时,观察到荧光强度显著降低,这与iPSC和NPC培养物中的观察结果相反(图2I-K)。虽然出乎意料,但这种与干细胞和祖细胞培养物相反的荧光强度变化趋势,与培养2周神经元的观察结果一致一一当时未发现显著差异。在对照组神经元培养物中进行的过氧化氢处理被用作氧化应激的阳性对照(补充图6B),我们证实CM 染料在ATM缺陷型和基因校正系中的氧化和线粒体螯合过程均呈线性关系(补充图6C),这意味着荧光强度的降低并非由于染料摄取达到平台期。单个OXPHOS复合物的蛋白表达水平显示,AT32突变神经元中复合物II的蛋白水平显著升高,其他复合物虽有变化但未达到显著性阈值 (补充图6D-F)。
虽然CM 检测细胞活性氧水平的方式是在染料氧化后才发出荧光,但线粒体的截留作用取决于膜电位。我们推测,神经元中ATM的缺失会导致线粒体膜电位长期受损(如TMRE所示),从而阻碍突变神经元有效摄取CM ,为验证这一假设,我们测量了ATM抑制对原本健康培养体系的影响。在培养4周进行线粒体评估前,对对照组神经元使用ATM激酶抑制剂(KU-55933或KU-60019)处理,并检测了线粒体含量(图3A-C)、膜电位(图3D-F)和氧化应激水平(图3G-I)。KU-60019导致MitoTracker深红染料的胞质荧光强度显著增强,且定位在神经突中的线粒体荧光斑点更加明亮并呈现聚集形态,这提示存在线粒体融合或线粒体自噬减少,与既往报道一致(Valentin-Vega等,2012)。支持这一发现的是,ATM抑制后定位在神经突的线粒体斑点显著减少(图3J)。急性ATM抑制后,胞质和神经突线粒体的TMRE荧光均有所增强(图3D-F)。与ATM缺陷神经元因线粒体膜电位长期受损导致CM 荧光减弱的观察结果相反,我们发现对具有健康线粒体的对照组神经元进行急性ATM化学抑制,会导致定位在胞质和神经突的线粒体中CM-H2Xros荧光显著增强(图3G-I),表明存在大量ROS产生。值得注意的是,根据细胞核计数,ATM抑制剂处理并未减少神经元数量(图3K),提示ATM信号缺陷在无细胞毒性作用的情况下驱动了线粒体异常。这些数据表明,缺乏功能性ATM信号通路的神经元中存在显著水平的氧化应激,支持了ATM作为氧化应激传感器及细胞ROS反应协调者的作用 (Guo等,2010)。这进一步凸显出ATM化学抑制可能与基因缺失不具有可比性。在所有情况下,ATM的缺失一一无论是由于ATM缺失的持续时间(急性或慢性),还是由于机制(化学抑制或基因突变)一一都会导致 。
我们共同得出结论:ATM缺陷神经元表现出严重的线粒体功能障碍,这种障碍在成熟过程中愈发明显,表现为线粒体含量或定位的持续改变,以及线粒体膜电位的降低一一这些变化可能影响呼吸链功能并加剧氧化应激水平(Amo等,2011)。
¶ 3.4.AT患者来源神经元及皮质类器官的转录组分析揭示氧化应激与线粒体缺陷
为了进一步探究这种线粒体功能障碍的可能机制和后果,我们对AT32突变型和同基因校正型iPSC、二维神经元培养物(2周龄)及三维脑类器官(100日龄)进行了批量RNA测序。我们证实突变型与校正型神经元具有相似的神经分化能力(附图7),通过对比AT32校正型与突变型神经元,鉴定出1836个差异表达基因(图4A);相比之下,脑类器官的差异表达基因数量接近9000个(图4B)。主成分分析对此作出解释:在基因集空间分析中,ATM缺陷型神经元和类器官与ATM正常型对应物的转录组可明确区分(附图8A、9A)。约 的神经元差异表达基因在脑类器官数据集中同样被识别(附图8C)。KEGG通路分析显示AT32突变型脑类器官存在显著的线粒体功能损伤,包括氧化磷酸化和TCA循环通路上调,以及参与ROS代谢、神经变性和细胞衰老的基因编码mRNA增加。相反,涉及突触功能、轴突导向和细胞周期信号通路的基因编码mRNA则出现下调(图4C)。GO术语分析显示氧化还原、细胞呼吸、电子传递链过程,以及包括ROS、氧化和内质网应激在内的应激反应过程显著富集,同时细胞与神经元死亡及调亡过程增加(图4D)。与ATM蛋白功能失活一致,DNA和双链断裂修复过程下调,神经发生、神经系统发育及突触传递相关过程也受到抑制。值得注意的是,线粒体与细胞器组织、分裂、定位和运输过程同样受到影响,这与我们观察到的线粒体在神经元胞体和突起间定位改变的结果相符(图4E)。所有类别的TOP10-15GO术语和KEGG通路详见附图8和9。
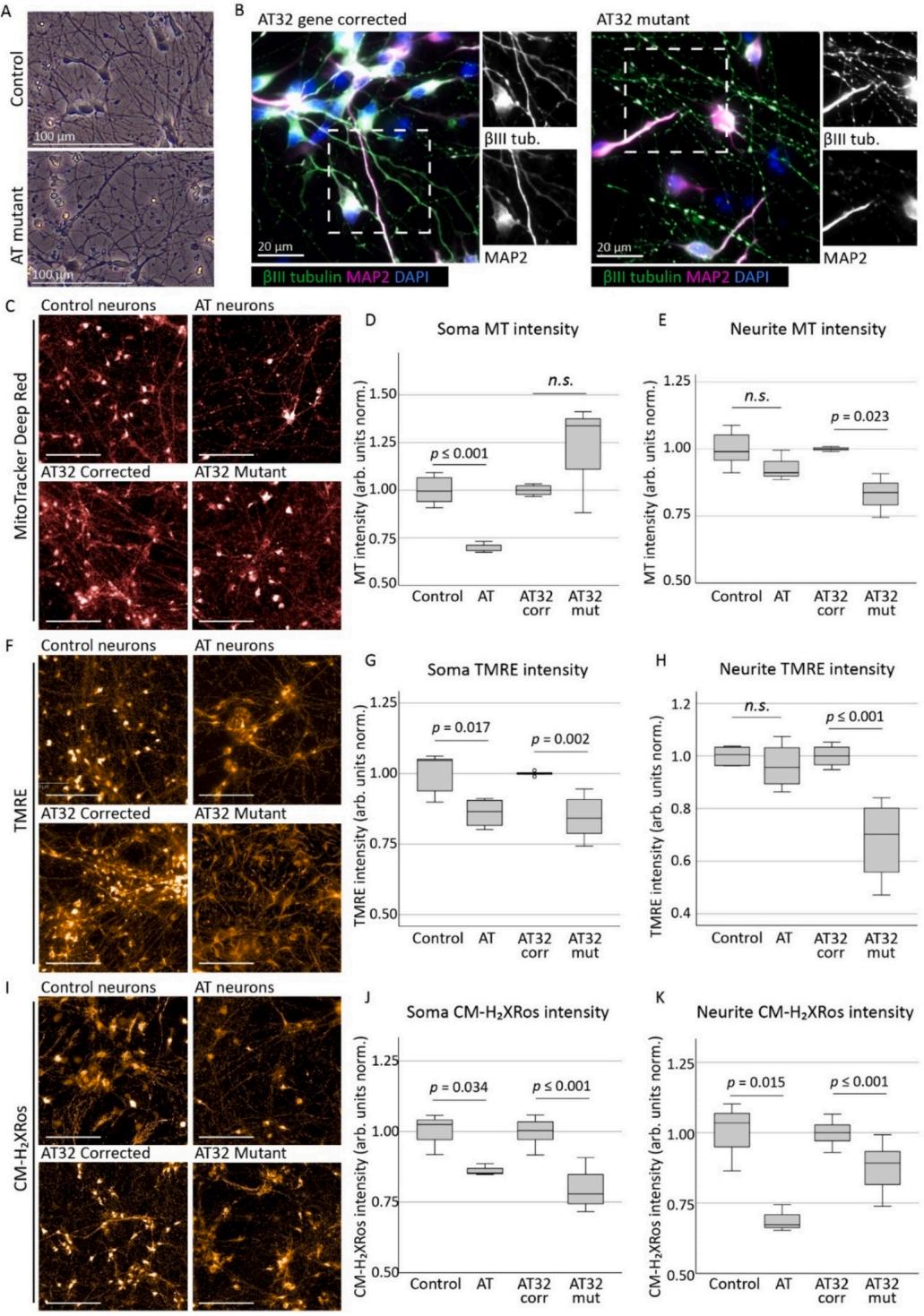
图2.AT患者iPSC衍生神经元的线粒体评估。A)由AT患者及对照/基因校正iPSC生成的神经元网络在PLO层粘连蛋白上培养并成熟2-4周。B)神经元表达神经标记物II微管蛋白和MAP2,AT突变神经元呈现与神经退行性表型一致的分段化特征。C)通过MioTacker深红染料评估线粒体含量,胞体荧光强度(D)显示AT神经元较对照组显著降低( ,但AT32神经元无此现象 ,相反,神经突荧光强度(E)显示AT32突变神经元投射区域的线粒体定位显著减少(t(4) ,而AT株系与非亲缘对照组无差异 TMRE检测(F)表明ATM缺陷神经元在4周时线粒体膜电位持续受损,尤以胞体线粒体为著(G;AT组对照 ;AT32同基因株系t( 。AT32突变神经元的神经突TMRE荧光强度(H)亦下降( ,但AT株系与非亲缘对照组无差异 。CM 染色(I)显示AT(t 与AT32神经元( 的胞体荧光强度(J)及神经突强度(K;分别为t ( 4 ) = 4 . 0 8 5 , p = 0 . 0 1 5 \Dot { \ast } \mathbb { H } ( 3 5 ) = 5 . 8 2 2 , p \leq 0 . 0 0 0 ) 较对应对照组均降低。标尺 箱线图显示中位数 四分位距,须线表示数据极值。(关于图例中颜色引用的解释,请参阅本文网络版。)
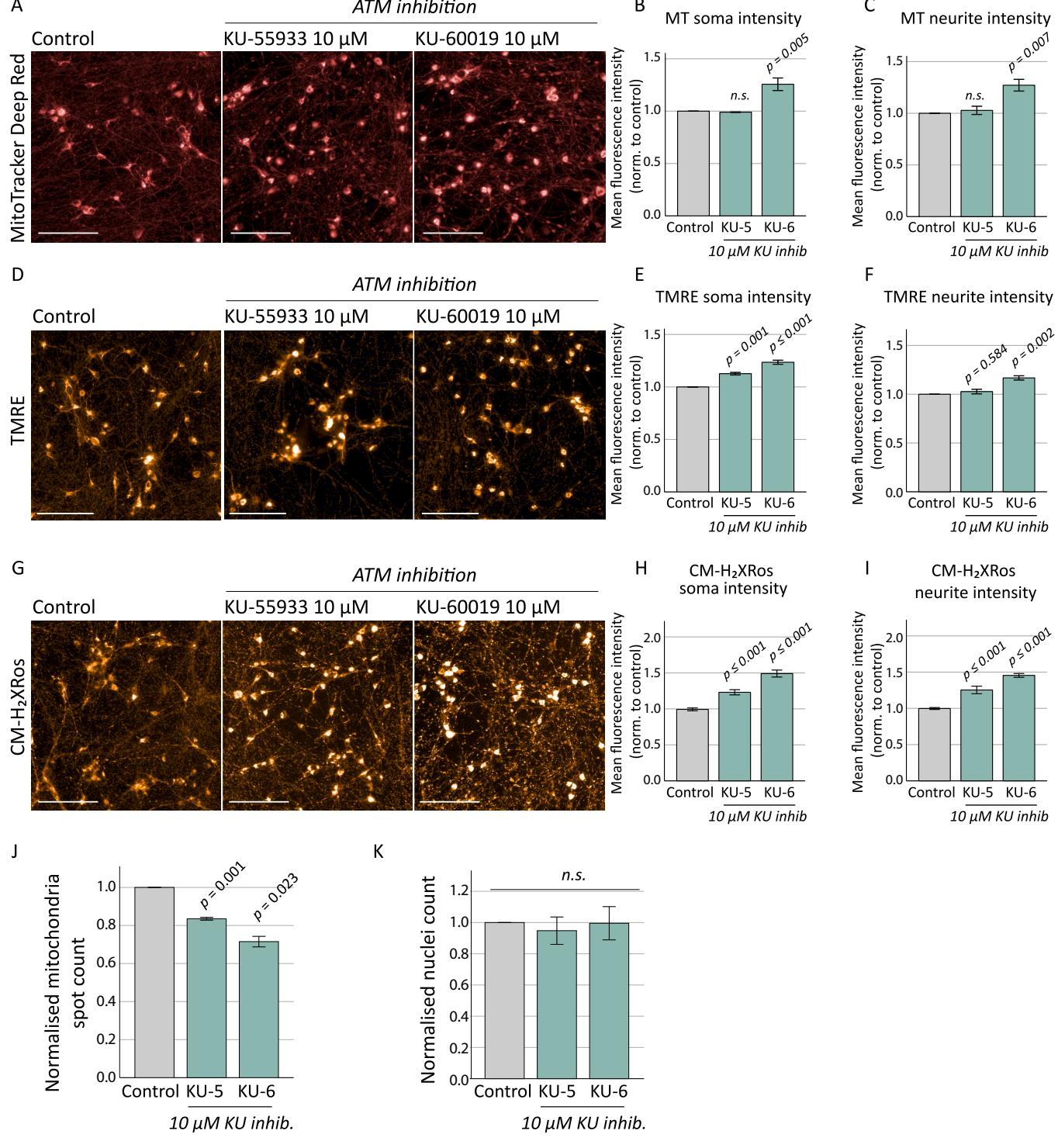
图3.经化学性ATM抑制剂处理的对照组神经元线粒体评估。对照组神经元在4周龄进行线粒体评估前,使用ATM抑制剂KU-5933和KU-60019( )处理10天。通过MitoTracker深红染色(A)显示,KU-60019处理引起胞体[B; 与神经突斑点[C; ]荧光强度增强,经ANOVA检验及Tukey事后分析确定对应p值分别为0.005与 KU-55933未引起线粒体含量显著变化。D)TMRE染色检测膜电位显示,KU-55933与KU-60019处理后胞体及神经突荧光强度均增加。E)胞体TMRE强度: $\mathrm { F } ( 2 , 8 ) { = } 7 8 . 2 8 9 , p { 0 . 0 0 1 $ ;Tukey事后分析显示KU-55933p 、KU-60019p (相较于对照组)。F)神经突TMRE强度:F(2,8)=23.535, ;Tukey事后分析显示KU-55933p 、K G)CM 荧光在ATM信号化学抑制后增强,表明显著氧化应激表型。H)胞体CM 荧光强度: ;Tukey事后分析: 、KU-60019p I)神经突CM 荧光强度: ;Tukey事后分析:KU-55933p 、KU-60019p J)ATM化学抑制导致神经元突起内线粒体斑点数量显著减少[F ,其中KU-55933( 与KU-60019 )均有效应,但ATM抑制未改变基于细胞核计数的神经元数量[K; 。抑制剂处理组均以未处理对照组为基准进行标准化。标尺: 误差线表示均值 标准误。KU-5:KU-55933;KU-6:KU-60019。(关于图例中颜色标注的说明,请参阅本文网络版。)
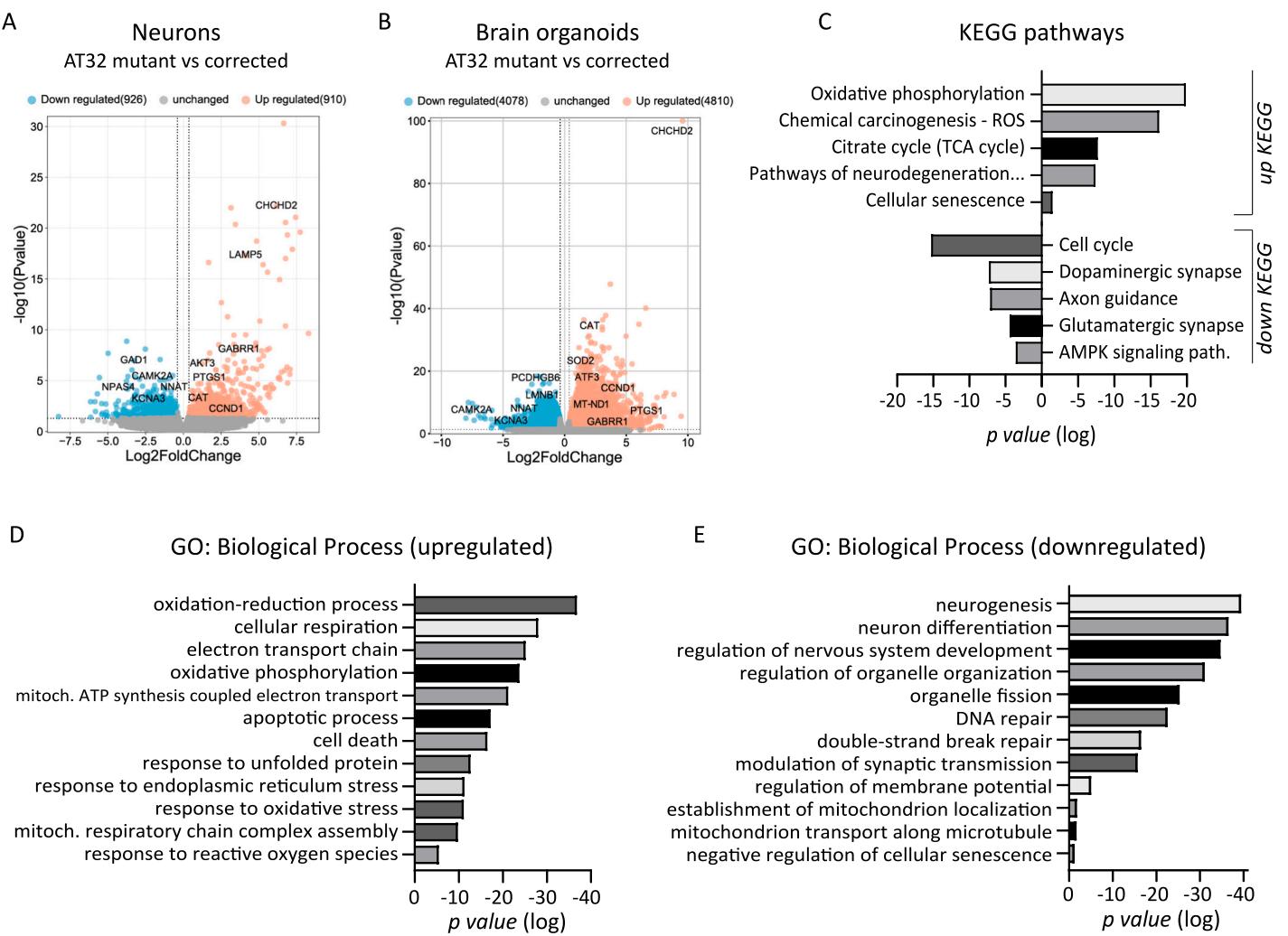
图4.AT32神经元与脑类器官转录组分析。对培养2周的AT32神经元和100天龄脑类器官进行RNA测序。A)火山图显示AT32突变型与基因校正型神经元对比鉴定出1836个差异表达基因(与校正型相比,突变型中910个基因上调、926个基因下调),而100天龄脑类器官则出现近900个差异表达基因(B;与校正型相比,突变型中4810个基因上调、4078个基因下调)。分别对上调与下调差异表达基因进行KEGG通路(C)和GO分析(D、E),发现这些基因在线粒体功能相关通路(如氧化磷酸化与电子传递链)以及应激响应过程(包括氧化应激与内质网应激)中显著富集。DEG:差异表达基因;GO:基因本体。
我们接下来通过分析氧化应激标志物的基因表达模式(图5A),研究了神经元和类器官中氧化应激的程度。与 相比,AT32突变神经元和类器官均表现出明显的氧化应激表型基因校正对应模型中,类器官模型里达到显著性的标记基因数量更多,这与我们早先关于线粒体功能障碍随神经元成熟度增加而加剧的结论一致。在ATM突变神经元和类器官中均显著上调的基因中,最值得关注的是CHCHD2((9.6 Log2FC, ),该基因也被称为MNRR1),是氧化应激与细胞应激反应(Purandare等,2018;Aras等,2013)、线粒体代谢、电子传递链(Aras等,2015)以及线粒体生物发生与形态调控(Aras等,2020;Liu等,2020)的关键调节因子。CHCHD2还能作为细胞凋亡抑制剂(Liu等,2015;Liu和Zhang,2015),并与神经退行性变及帕金森病相关(Kee等,2021;Jansen等,2015;Ikeda等,2022;Ikeda等,2019;Funayama等,2015;Imai等,2019)。在ATM突变神经元和类器官中上调的其他重要氧化应激基因包括超氧化物歧化酶SOD2 和SOD3,PTGS1、(环氧化酶1(COX-1))、CAT(过氧化氢酶)以及SQSTM1(p62)。过氧化氢酶和p62通过ATM磷酸化PEX5介导过氧化物酶体自噬,这一过程能招募p62并引导自噬体靶向过氧化物酶体,从而响应活性氧诱发pexophagy(Zhang等,2015)。既往研究已在过氧化物酶体中发现ATM存在(Watters等,1999),其含有的FATC结构域被认为是过氧化物酶体靶向序列。值得注意的是,患有该FATC结构域的突变并未表现出AT患者典型的放射敏感性(Guo et al.,2010),表明该结构域的功能独立于ATM的DNA损伤应答。
为间接评估神经元活性,我们检测了即刻早期基因的表达情况一一这类基因已知依赖于功能性线粒体。发现在ATM突变神经元培养物中NPAS4、FOSB、FOS和NR4A2的表达显著降低(图5A),这与突触传递及功能相关KEGG和GO条目的下调趋势一致(图4)。这种现象在类器官模型系统中并不明显,可能因为类器官包含大量其他细胞类型,从而掩盖了神经元基因表达水平的细微变化。我们还评估了衰老和炎症标志物,因先前研究表明AT脑类器官相较于胚胎干细胞衍生对照组具有更高的衰老程度(Aguado等,2021)。与这些数据一致,在培养100天的ATM突变类器官中,相较于同基因对照组也观察到明显的衰老表型。
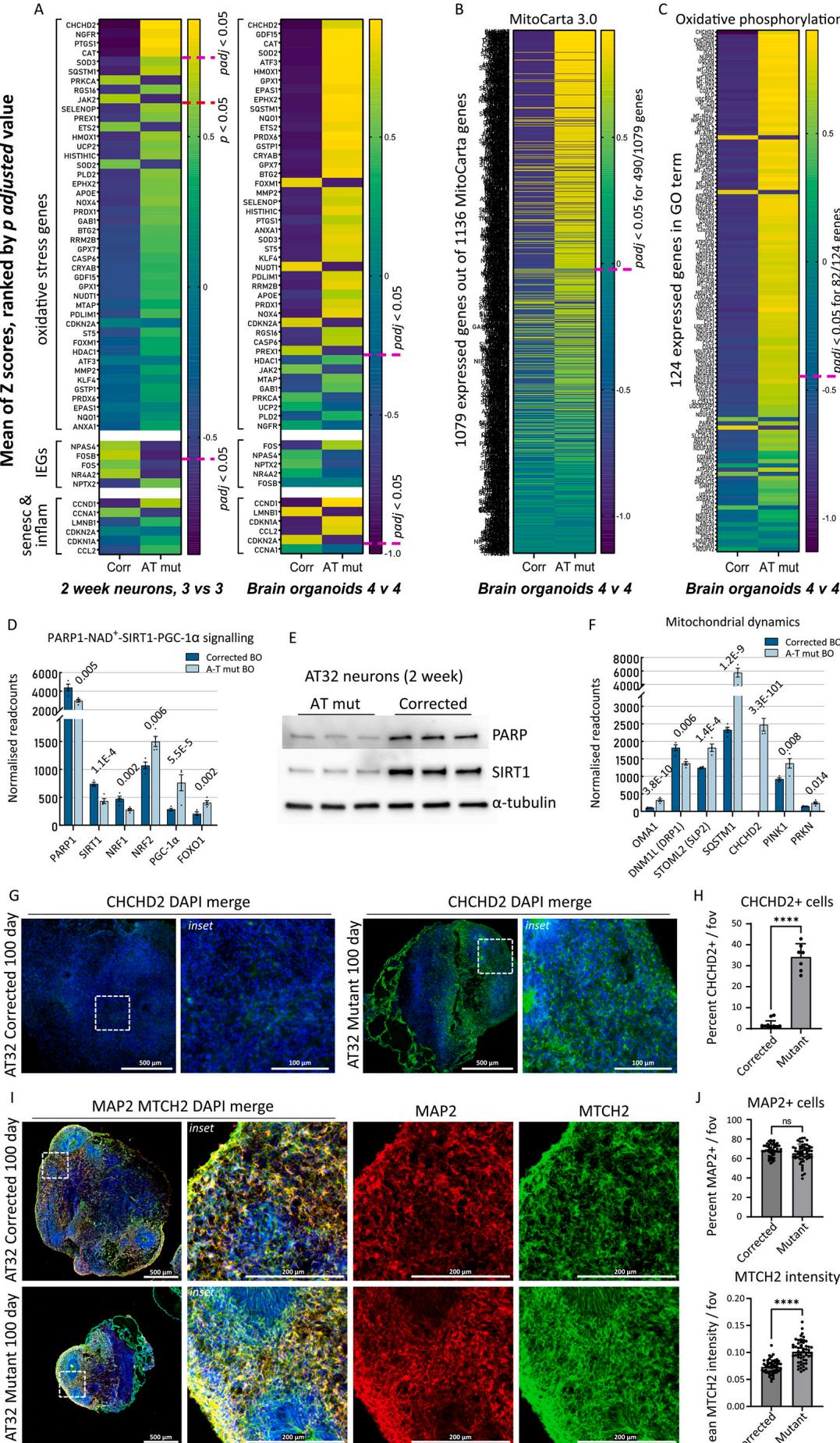
Figure5-artboard extended to 3 6 8 \mathsf
图5.AT32神经元和脑类器官中揭示的氧化应激特征与线粒体紊乱。
A-C)根据AT322周龄神经元和10天龄脑类器官RNA测序数据计算出的平均Z值,以热图形式展示AT突变型和校正型,基因按校正后的p值排序。粉色虚线标示padj 0.05的临界值。A)通过对比突变型与校正型神经元及脑类器官,绘制了氧化应激标志物、即刻早期基因以及衰老与炎症标志物的表达谱。B)所有表达的Mitocart 3.0基因的平均Z值在AT突变型与校正型脑类器官中的对比展示,C)同时展示了包含GO术语“氧化磷酸化”的所有表达基因。D)脑类器官RNA测序数据中PARPl-SIRT1信号通路相关基因的标准化读数,包括PARP1、SIRT1、NRF1、NRF2(NFE2L2)、PGC-1Q(PPRGC1A)及FOXO1。标注相应padj值。E)3个独立AT322周龄神经元培养物全细胞提取物中PARP和SIRT1表达的蛋白质印迹分析,以 -微管蛋白作为上样对照。F)脑类器官RNA测序数据中裂变/融合及线粒体自噬相关基因的标准化读数,包括OMA1、DNMIL(DRPI)、STOML2(SLP2)、SQSTM1(p62)、CHCHD2、PINK1和PRKN。标注相应padj值。G)AT32突变型与基因校正型脑类器官切片CHCHD2染色的代表性免疫荧光图像。比例尺 ,插图 H)AT32突变型类器官CHCHD2阳性细胞百分比显著升高[ ,数据来自5个脑类器官的7-10个视野。I)AT32突变型与基因校正型脑类器官切片MTCH2和MAP2染色的代表性免疫荧光图像。比例尺 ,插图 J)MAP2阳性细胞百分比无显著变化[t(10 ,而AT32突变型类器官中MTCH2强度增强[ ,数据来自5个脑类器官的50-55个视野。误差线表示均值±标准差。B0:脑类器官;FOV:视野;Corr:AT32基因校正型;Mut::AT32突变型;IEG:即刻早期基因;GO:基因本体。(关于图例中颜色引用的解释,请参考本文网络版。)
通过公正评估,我们分析了MitoCarta3.0目录中所有基因的平均表达量(图5B)。该目录全面收录了1136个编码线粒体定位蛋白的人类基因。我们还分析了包含氧化磷酸化GO术语的所有表达基因(图5C)研究显示ATM突变体存在显著的线粒体功能损伤,在1079个表达的MitoCarta3.0基因中,约半数基因表达增强,其中涉及氧化磷酸化的基因有三分之二上调,尤其是泛醇-细胞色素C还原酶(UQCR个基因)和NADH-泛醌氧化还原酶亚基(MT-ND、NDUF个基因)。这些基因的持续上调使我们推测其是对NAD+水平长期降低的代偿机制,这与既往在AT缺失小鼠中观察到NAD+(耗竭的现象一致(Fang等,2016))。这种NAD+缺乏位于聚(ADP-核糖)聚合酶1(PARP1)下游,导致SIRT1失活并引发线粒体自噬缺陷及线粒体功能障碍。因此我们在ATM突变模型中研究了PARP1-SIRT1信号通路,并纳入ATM磷酸化靶标核呼吸因子1(NRF1)(Chow等,2019)以及受活性氧和/或ATM调控的NRF2(Navrkalova等,2015;Lee和Paull,2020),二者均是线粒体生物发生和抗氧化功能的关键转录因子(Scarpulla,2008),且与PARP1、SIRT1及转录共激活因子PGC-lα、FOXO1存在相互作用(Hossain等,2009;Bai等,2015)。发现ATM突变类器官中PARP1、SIRT1和NRF1的mRNA水平降低,这与既往在ATM缺失细胞中观察到的NRF1蛋白减少一致(Chow等,2019),而NRF2、PGC-1α和FOXO1表达升高(图5D)。蛋白质印迹分析证实这些突变神经元中PARP1和SIRT1蛋白水平显著降低(图5E)。我们进一步推测线粒体网络动态上游调控因子(CHCHD2、PARP1、PGC-1α)的变化会对控制裂变/融合及线粒体自噬的基因产生下游影响。确实,我们发现了关键裂变基因OMA1、DNM1L (DRP1)和STOML2(SLP2)的紊乱(Wai和Langer,2016),以及线粒体自噬基因SQSTM1(p62)、PINK 和PRKN(的显著上调(图5F,附图10A))。随后我们证实ATM突变类器官中CHCHD2的上调表达体现在蛋白水平,观察到AT32突变类器官切片中CHCHD2免疫荧光强度较同基因对照显著增强(图5G、H)。通过用线粒体外膜蛋白MTCH2对AT32类器官切片进行免疫标记(图5I),我们评估了突变类器官中线粒体基因的转录上调是否与线粒体含量改变相关。突变类器官中MTCH2强度显著增加,而MAP2阳性细胞百分比未发生改变(图5J)。
¶ 3.5.AT患者来源的神经元模型呈现与氧化应激及线粒体功能障碍相关的炎症和衰老特征
AT病理学的一个重要方面且与线粒体功能障碍高度关联的是细胞衰老;它们共同构成了与AT相关的衰老标志中的两个(Aguado等人,2022年)。线粒体功能障碍是细胞衰老的普遍特征和驱动因素(Wiley等人,2016),其异常分裂与融合、过量活性氧产生、促炎分泌表型的形成以及线粒体代谢物(尤其是NAD)失衡均被证实会促进衰老表型(Martini和Pass0s,2023;Miwa等人,2022)。我们此前已发现ATONS-iPSC来源的脑类器官相较于无关对照组出现早发性衰老(Aguado等人,2021),因此我们进一步探究了AT32突变型与同基因型类器官的衰老及炎症程度。
KEGG通路分析(图4)显示ATM突变型脑类器官中细胞衰老增加,我们分析了构成细胞衰老GO术语的所有转录基因的平均表达量 (图6A)。组成该GO术语的基因中近半数呈现显著失调。脑类器官标准化读数评估识别出经典的衰老标志物,包括突变型BO中CCND1、CDKN1A(p21)和CCL2水平升高,而LMNB1表达降低(图6B)。2周龄神经元培养物未呈现显著差异(图5A)。为探究神经元进一步成熟后是否会显现衰老特征,我们培育了AT32突变型及修正型神经元并使其成熟10周。经过延长期成熟后,检测到AT突变型神经元中呈现衰老相关β-半乳糖苷酶染色的细胞比例增加3倍(SA -Gal;图6C、D)。通过免疫化学检测发现衰老神经元培养物中p16和p21表达显著升高,证实AT突变型神经元的衰老水平有所增强(图6E-G)。
¶ 3.6.抗氧化治疗可纠正氧化应激并恢复神经元功能
为评估氧化应激位于线粒体功能障碍的上游还是下游,我们在测定氧化应激水平前,用抗氧化剂N-乙酰半胱氨酸(NAC)处理AT32突变神经元10天。NAC处理使胞质内CM 信号降低约 (图7A、B),但未能改善线粒体膜电位(图7C)。通过对AT32校正组、突变组及NAC处理突变神经元的微管蛋白免疫标记,我们测量了神经突延伸、分支和存活情况(图7D)。NAC处理显著挽救了ATM缺陷神经元的神经突生长,使突变神经元的视场覆盖率恢复至与校正神经元相当的水平(图7E)。为验证NAC和/或回补性化合物庚酸(C7)是否能使AT神经元中失调关键基因的表达水平正常化,我们分离RNA进行基因表达分析。与未处理组相比,NAC处理的AT神经元中CHCHD2mRNA表达降低,而TUBB3 mRNA未发生变化(图7F)。处理组AT神经元中SOD3、NPAS4、SYP及SLC17A7 (VGLUT1)的RNA表达水平虽呈现向校正组靠拢的趋势,但未达到显著性水平,表明可能需要更长的治疗周期来纠正这些特征(附图1OB)。
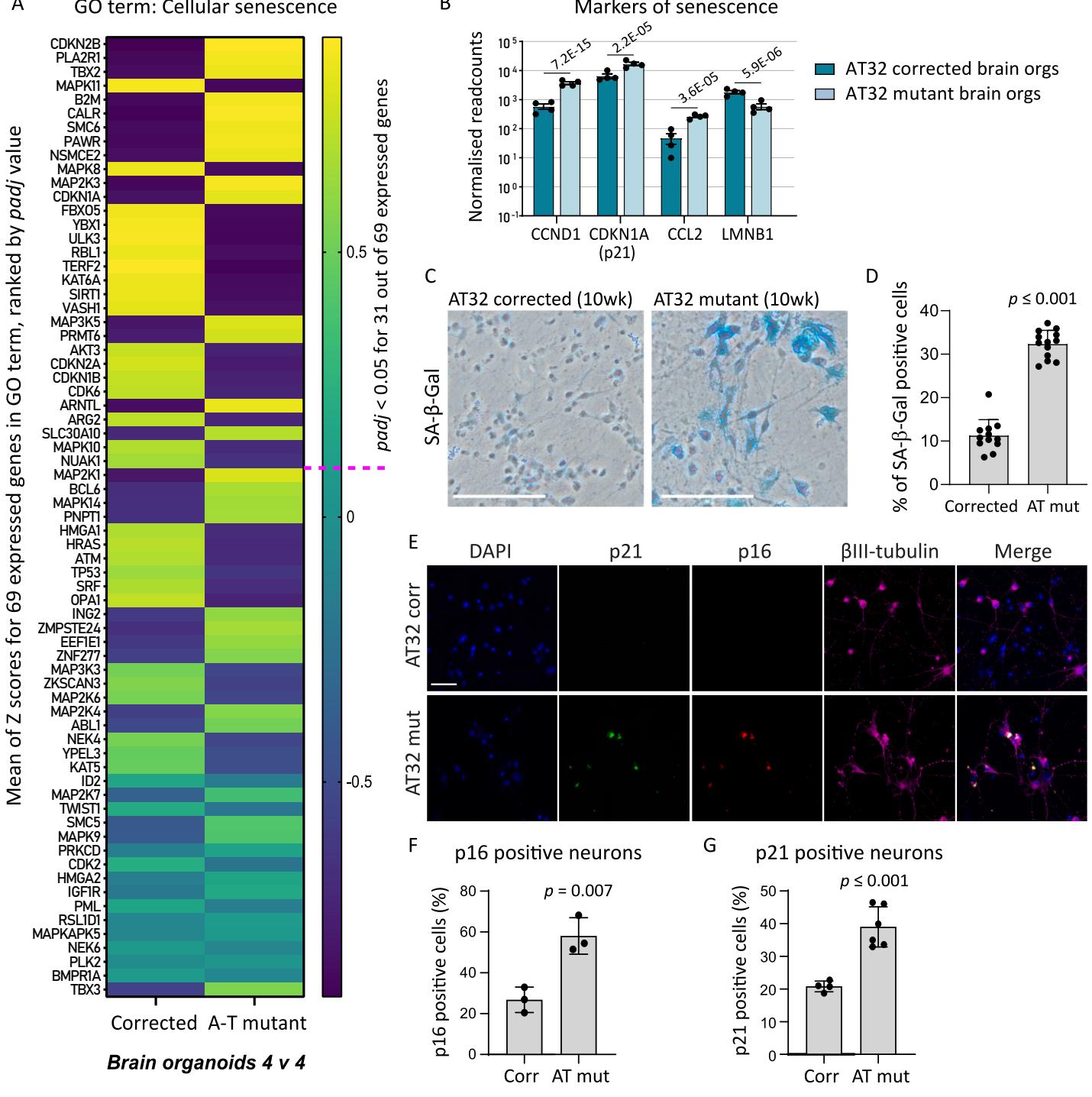
图6.AT32神经元和脑类器官中揭示的衰老特征。A)包含GO术语"细胞衰老"的所有表达基因的平均Z值通过热图展示,数据来源于AT32脑类器官RNA测序。基因按校正后的p 值排序,粉色虚线标示padj 的临界值。B)脑类器官RNA测序数据中衰老标志物的标准化读数计数,padj 值如图所示。C)AT32突变型与校正型10周龄神经元进行SA -gal检测的代表性图像。比例尺 D)AT32突变型神经元的SA-β-gal阳性染色比例显著更高[ ,数据来自3次独立分化的12个视野。E)经p21和p16染色的AT32突变型与基因校正型10周龄神经元的代表性免疫荧光图像。比例尺 F)p16阳性神经元定量[t(4 与G)p21阳性神经元定量 ,均来自3次独立分化实验。误差线表示 标准差。FOV:视野;Corr:AT32基因校正型;Mut:AT32突变型;GO:基因本体。(关于图例中颜色标注的解释,请参阅本文网络版。)

图7.抗氧化剂处理可降低氧化应激并恢复神经元放电。
A)AT32突变神经元在测量氧化应激水平前,用抗氧化剂NAC处理10天,氧化应激水平通过CM s检测。标尺 B)NAC处理后CM 荧光强度降低;t值(Bakkenist和Kastan, , ,而TMRE荧光强度(C)在处理后的神经元中保持不变; , 误差棒表示 标准误。D)3组独立的AT32校正型、突变型及NAC处理突变型神经元培养物的代表性二元βII-微管蛋白免疫化学染色。E)通过量化二元βII-微管蛋白免疫化学染色计算每个视野的覆盖率百分比; , ,经Tukey事后分析发现突变型神经元相较于校正型显著降低! ,且NAC处理可显著纠正此表型 )。误差棒表示士标准误。F)从AT32突变型和校正型神经元中提取总RNA,用于量化CHCHD2和TUBB3.mRNA表达水平。突变型神经元培养物分别经NAC或C7处理。ETFA作为内参基因。ANOVA[F $p \le 0 . 0 0 1 $ 及Tukey事后分析显示CHCHD2mRNA水平存在显著差异, 值如图所示。TUBB3 mRNA表达保持不变[F , ,3次独立重复实验,误差棒表示 标准差。G)对上下调差异表达基因分别进行GO分析,发现突触功能与神经递质信号通路显著下调。H)通过MEA平台将脑类器官固定于电极芯片,记录基线电活动后加入谷氨酸。计算谷氨酸刺激后时间窗口(4、12、24、48秒及刺激后总时长)的平均放电率(每秒峰值数)并标准化至基础放电率。采用学生t-检验判定显著性, 值如图所示。校正型类器官:4个MEA芯片上6个类器官的2834个活跃电极;突变型:3个MEA芯片上5个类器官的4510个活跃电极。误差棒表示 倍标准误。I-J)AT32突变脑类器官经C7和NAC处理两周后固定于电极芯片,记录基线电活动后加入谷氨酸。计算基线及谷氨酸刺激后标准化至基线的平均放电率(每秒峰值数)。(I)采用Welch’s ANOVA分析基线神经元活动[F , ,Games-Howell事后分析显示突变型基线放电较校正型显著降低 )。(J)谷氨酸刺激显示未处理与处理突变型类器官间存在显著差异[F(3,6435] , ,Games-Howell事后分析如图所示]。误差棒表示 倍标准误。GO:基因本体;MEA:多电极阵列;NAC: 乙酰半胱氨酸;C7:庚酸盐。
鉴于AT患者iPSC衍生神经元表现出即刻早期基因表达降低,且我们的AT类器官RNAseq数据中突触和神经递质通路KEGG/GO条目显著富集(图4、5,图7G重点标注),我们决定测试神经元放电在多重电极阵列平台上,100天龄类器官于抗氧化处理存在及缺失条件下的电生理记录能力。结果显示,AT32突变类器官对谷氨酸刺激的反应显著减弱(图7H),且在静息(未受刺激)状态下平均放电频率较基因校正对照组明显降低(图7I)。这种AT突变类器官对谷氨酸的反应能力与GO术语转录组分析结果一致,该分析发现谷氨酸受体信号通路在突变类器官中显著下调(图7G)。研究团队用抗氧化剂NAC以及C7处理脑类器官—-C7能恢复TCA循环活性,并被证明可缓解代谢应激条件下ATM-HBEC和AT患者来源ONS细胞的线粒体功能障碍与细胞死亡(Yeo等人,2021b)。经C7和NAC处理的脑类器官在未受刺激状态下未显示神经元放电频率的改善(图7I),但在谷氨酸刺激下,经NAC与C7处理的AT突变类器官平均放电频率较未处理组显著提升,其中NAC处理组的放电频率甚至恢复至基因校正类器官的水平(图7J)。
¶ 4.讨论
ATM是线粒体稳态和氧化应激的重要主调控因子,这一点已通过小鼠模型(Kamsler等人,2001;Quick和Dugan,2001;Chen等人,2003)和人类患者细胞(Reichenbach等人,2002;Ambrose等人,2007;Valentin-Vega等人,2012)的研究得到证实。尽管如此,患者神经元细胞中线粒体功能障碍和氧化应激的程度仍未得到明确表征。本研究首次利用患者原代细胞和iPSCs,在从干细胞、神经元前体到二维神经元培养体系,最终到脑类器官的不同成熟阶段的人类神经模型中,系统检测了线粒体含量、膜电位和氧化应激水平。我们的发现对于理解AT神经病理学具有特殊重要意义,特别是考虑到当前体内模型并未表现出在AT患者中持续观察到的关键神经异常(包括神经退行性病变)(Lavin,2013)。
我们首先检测了5名AT患者与对照组相比,嗅觉上皮来源的ONS细胞的线粒体含量、膜电位和ROS产生情况,未观察到疾病特异性差异。由于ATONS细胞表现出辐射超敏性、辐射诱导信号传导缺陷及细胞周期检查点缺陷(Stewart等,2013),我们的数据表明ATM缺陷ONS细胞中的线粒体损伤和氧化应激并不仅仅是核DNA损伤累积的下游结果(Guo等,2010;Chen等,2023)。随后我们利用其中两名AT患者来源的iPSC,在从干细胞、祖细胞到脑类器官的神经元成熟过程中监测线粒体功能。我们观察到显著的线粒体功能障碍和氧化应激水平,且随着神经元模型成熟度的提高,这些异常表现逐渐加剧。
¶ 4.1.AT患者神经元表现出线粒体膜电位缺陷及氧化应激增加
线粒体膜电位由氧化磷酸化过程中的质子泵产生,主要反映电子传递链活性,是线粒体功能和细胞能量状态的指标。与对照/等基因校正神经元相比,我们在AT患者来源神经元中观察到线粒体膜电位显著降低,提示线粒体功能下降。支持这一结论的是线粒体内CM-H2Xros积累的逆转。虽然iPSC和NPC培养物中CM-H2Xros荧光增强与先前观察到的氧化应激水平升高一致(Ovchinnikov等,2020),但它们在2周时恢复至无显著性差异,随后在4周时水平下降的现象最初令人困惑。然而,该染料在线粒体内的聚集取决于膜电位,我们得出结论:在神经元模型中揭示的长期电位丧失,结合线粒体稳态失衡和定位异常,解释了CM 强度降低的原因。将AT患者来源神经元与经ATM抑制剂处理的对照神经元进行比较进一步支持该理论,因为在其他方面健康的神经元模型中进行急性ATM抑制并不会降低膜电位,反而显著增加线粒体内CM-H2Xros的积累,表明在缺乏ATM激酶活性时产生了显著活性氧,这与既往研究证实这些抑制剂会阻断ATM对氧化应激的反应相一致(Guo等,2010;Zhan等,2010)。抗氧化治疗被证明可降低ATM缺陷神经元的氧化应激并促进神经突生长和存活,但对膜电位无影响,提示氧化应激是线粒体功能和/或膜电位受损的下游过程。神经元及脑类器官的转录组学分析进一步证明了强烈的氧化应激表型,该表型在类器官中更为显著,表明氧化应激随模型成熟度和复杂性的增加而加剧。
¶ 4.2.线粒体稳态过程(包括定位、分裂-融合、线粒体自噬和生物发生)在AT患者神经元及类器官模型中均出现异常。
线粒体本质上是高度动态的,它们形成相互连接的网状结构,并沿着神经元突触主动迁移至能量需求较高的区域。它们持续进行严格调控的裂变与融合循环,以维持功能性线粒体群体的稳定,这凸显了线粒体健康在神经系统中的重要性(Seager等人,2020年)。我们观察到 之间的差异在神经元成熟过程中,AT神经元与同基因对照组的线粒体含量及神经元突起内定位趋于一致一一培养两周时,胞体线粒体减少与神经元投射区线粒体增加同步出现,表明线粒体向突起区域转移,这可能是维持神经突和突触完整性的代偿机制。培养至4周时,这种分布模式消失,转而呈现整体性减少。值得注意的是,虽然线粒体总量减少,但神经突内线粒体斑点数量保持稳定或增加,说明随着时间推移功能性ATM蛋白复合物缺失,导致线粒体分裂时结构动力学改变,最终形成碎片化线粒体网络。这种现象与其他神经退行性疾病及衰老相关病理改变一致(Wai和Langer,2016;Mollo等,2020),也与Luo、Lyu等人(Luo等,2023)的观察结果相符一一他们发现ATM抑制会加剧线粒体过度分裂。
近期研究发现,ATM缺陷小鼠及ATM缺失细胞中存在线粒体自噬和线粒体裂变受损,以及PARP1下游NAD+/SIRT1信号通路减弱的现象(Fang等,2016;Fang等,2014)。补充NAD+被证实可改善线粒体自噬与DNA修复,并减轻共济失调毛细血管扩张症神经病理的严重程度(Fang等,2016)。值得注意的是,我们在AT患者神经元模型中观察到PARP1和SIRT1的转录水平及蛋白表达均有所下降。PARP1与SIRT1均为核酶,共同以NAD+为底物,并相互抑制彼此活性。PARP1激活会快速导致线粒体膜电位丧失和活性氧产生,而较高的SIRT1活性则能改善线粒体功能、促进线粒体生物合成并抵御活性氧损伤(Bai等,2015;Singh等,2018;Xu等,2018)。研究已知在无细胞损伤情况下,ATM与PARP1可在体内形成分子复合物(Aguilar-Quesada等,2007)。除直接相互作用外,ATM还可能通过调控NRF1(线粒体生物合成基因的关键转录因子之一)来影响PARP/NAD+/SIRT信号轴。NRF1是ATM在氧化应激 (而非DNA损伤)激活状态下的磷酸化靶点,其磷酸化可促使NRF1核转位并上调线粒体及氧化磷酸化相关基因。人脑样本显示,与小脑其他神经元相比,NRF1在浦肯野神经元中表达更为富集,而AT患者脑样本中NRF1的核转位现象则有所减弱(Chow等,2019)。
综上所述,我们推测ATM可能通过直接结合PARP和/或通过NRF1调控PARP活性来调节PARP/SIRT稳态。NRF1和SIRT1通过激活FOXO1和PGC-1α来调节线粒体稳态与生物合成,而我们的AT患者脑类器官研究显示这些转录水平均出现紊乱。结合PARP/SIRT信号传导中断,负责线粒体分裂(OMA1,、DRP1,SLP2)和线粒体自噬(PCG-1α,SQSTM1,PINK1、PRKN)的关键信号通路失调,使我们得出结论:ATM是线粒体更新的核心调控因子,能通过多种途径施加影响,且功能性ATM蛋白的缺失直接导致分裂-融合平衡受损、生物合成增加以及线粒体自噬减少,最终引发受损线粒体的累积。
源自AT患者的脑类器官进一步证实了线粒体维持功能受损,AT类器官比同基因类器官含有显著更多的线粒体内容物,这表明除了线粒体运输和裂变-融合过程存在障碍外,线粒体更新和/或生物合成也存在严重缺陷。我们的转录组分析支持这一结论,其中观察到与线粒体动力学相关的基因表达发生改变。尤其值得注意的是CHCHD2 (MNRR1)的大幅上调一一该基因是OXPHOS和线粒体稳态的调控因子(Aras等人,2015)。在应激条件下,CHCHD2的线粒体导入被抑制,导致其在细胞核内积累,进而作为细胞色素c氧化酶亚基的转录因子发挥作用(COX)(Aras等,2013)。重要的是,研究发现CHCHD2的过表达可诱导线粒体未折叠蛋白反应、自噬和线粒体生物合成(Aras等,2020),这与我们在AT脑类器官中观察到的线粒体含量显著增加相一致。CHCHD2还可作为凋亡抑制剂(Liu等,2015;Liu和Zhang,2015);有趣的是,许多抗凋亡因子在衰老过程中会上调(Salminen等,2011),正如下文将讨论的,这已成为AT脑类器官的显著特征。
目前AT也被视为一种早衰性疾病,线粒体功能障碍是其衰老表型的关键特征。我们实验室此前已证明,与hESC衍生的对照类器官相比,AT脑类器官表现出强烈的衰老表型,而通过抑制cGAS-STING通路可逆转此现象(Aguado等,2021)。本文我们在100日龄的等基因AT及基因校正脑类器官、以及10周龄的二维神经元培养体系中验证了这一发现,这些模型均显示出增强的SA- -Gal染色及p16、p21蛋白表达。线粒体功能障碍常被忽视作为衰老和细胞衰老的标志(Aguado等,2022),大量研究表明线粒体动力学受损(如裂变周期减少)会促进衰老相关分泌表型(SASP)发展并增强抗凋亡能力,导致线粒体膨大和过度融合。值得注意的是,线粒体自噬缺陷也可能诱导细胞衰老(Martini和Passos,2023)。与我们发现PARPSIRT信号通路和线粒体自噬过程受损以及线粒体含量增加的观察结果相吻合,其他研究也证实补充NAD+能够促进AT成纤维细胞的线粒体自噬,且增强的线粒体自噬通过PINK依赖性方式阻断了STING介导的衰老进程(Yang等,2021)。补充NAD+还可抑制ATM/小鼠的神经退行性病变和衰老表型,并改善运动功能与线粒体稳态。基于这些发现,结合我们在AT神经元模型中观察到的加速衰老现象,我们得出结论:线粒体功能障碍是导致AT患者衰老表型的重要因素。
¶ 4.3.抗氧化剂或补给剂治疗可挽救ATM类器官中受损的神经元信号传导
ATM研究中一个较少被关注的方面是其在内囊泡和/或蛋白质运输机制中的作用,以及与突触囊泡的关联(Pizzamiglio等人,2020)。ATM可与VAMP2和SYNAPSIN-I结合,而ATM缺失会导致突触囊泡循环受损,表明胞质ATM在神经元活动中具有调节作用(Li等人,2009)。后续研究更精确定位ATM存在于兴奋性(VGLUT1)囊泡中(Cheng等人,2018)。相应地,在海马神经元培养物中降低ATM会导致兴奋/抑制平衡向抑制方向偏移,增加GABA能突触数量,并降低神经元兴奋性(Pizzamiglio等人,2016)。我们的研究结果强有力支持这些数据—一GO和KEGG通路分析均显示突触和囊泡运输相关条目减少,不过我们的大脑类器官模型中显著的线粒体损伤很可能也导致了神经元功能的下游改变。对100天龄大脑类器官的MEA分析显示,未受刺激/基础放电活动水平降低,神经元对谷氨酸刺激的反应能力减弱,而这种缺陷可通过NAC或C7处理得以缓解。这是首次在AT患者来源的大脑类器官中观察到功能性神经元缺陷的例证,表明虽然ATM可能在突触功能中具有抗氧化治疗难以逆转的机制性作用,但通过这些治疗增强线粒体健康并减少氧化应激,或许能支撑神经元活动。
总之,我们的数据表明AT患者神经元表现出渐进性氧化应激表型、线粒体膜电位受损、自噬失调以及裂变调节功能受损融合过程。我们发现了与维持线粒体动力学相关的基因表达模式改变,包括CHCHD2的显著上调和PARP/SIRT信号通路的改变,这些改变导致了线粒体缺陷。此外,我们在AT大脑类器官中观察到衰老水平升高和神经元活性降低,重现了AT的衰老和神经退行性特征。抗氧化剂治疗在功能上改善了AT大脑类器官和神经元,提高了神经元放电率并减少了活性氧。我们的研究首次系统性地描绘了非转化人类神经元细胞类型中线粒体功能障碍的性质和时间调控,并表明涉及线粒体稳态和抗氧化信号通路的多个ATM依赖途径受损,共同导致了AT的神经退行性病变。
¶ 资金信息
本研究得到了澳大利亚国家健康与医学研究理事会(申请号1138795、1127976、1144806和1130168)、BrAshA-T基金会、佩里·克罗斯脊柱研究基金会以及ARC探索项目(DP210103401)的资助。
¶ 作者贡献
J.A.完成了图6E、F所示的染色及分析工作,并制备了用于RNA测序的脑类器官。C.G.I.生成了图6G-I的结果。H.K.C.培育了人脑类器官。A.F.协助完成了测序分析。Z.H.完成了附图3所示的染色工作,并协助完成iPSC的生成、分化与培养维护。M.L.和A.M.S.提供了ONS细胞及学术支持。H.L.完成了所有其他数据的生成、分析与解读,设计了研究方案并撰写了论文。E.W.协助完成研究设计、数据解读、文稿撰写并提供了资金支持。所有作者均参与了论文的修改。
¶ CRediT作者贡献声明
汉娜·C利森:写作-审阅与编辑、写作-初稿撰写、可视化、验证、监督、项目管理、方法论构建、调研实施、形式分析、数据整理、概念化框架。胡里奥·阿瓜多:写作-审阅与编辑、调研实施、形式分析。塞西莉亚·G梅斯-因克安:调研实施、形式分析。哈曼·考尔·查格:方法论构建、调研实施。阿特法·塔赫里安·法德:形式分析。佐伊·亨特:方法论构建、调研实施。马丁·F拉文:资源供给、概念化框架。艾伦·麦凯-西姆:资源供给、概念化框架。恩斯特·J沃尔维唐:写作-审阅与编辑、写作-初稿撰写、监督指导、资源供给、资金获取、概念化框架。
¶ 利益冲突声明
作者声明,他们没有已知的竞争性财务利益或个人关系,这些可能影响本文报道的工作。
数据可用性数据将根据要求提供。
¶ 致谢
我们感谢生物医学科学院显微镜与影像分析中心(昆士兰大学)的Maria Kasherman和Shaun Walters提供的技术支持,以及E.W.实验室全体成员的讨论。我们向捐赠组织并热情支持我们研究的患者及其家属致以诚挚谢意。
附录A.补充数据
本文的补充数据可在 https://doi.org/10.1016/j.nbd.2024.106562 在线获取。
¶ 参考文献
Aguado, J., et al. (2021). 抑制 cGAS - STING 通路可改善共济失调 - 毛细血管扩张症脑类器官的早衰特征. Aging Cell, 20(9), e13468.
Aguado, J., et al. (2022). 共济失调 - 毛细血管扩张症的衰老特征. Ageing Res. Rev., 79, 101653.
Aguilar - Quesada, R., et al. (2007). DNA 损伤响应中 ATM 与 PARP - 1 的相互作用及通过 PARP 抑制对 ATM 缺陷细胞的增敏作用. BMC Mol. Biol., 8, 29.
Ambrose, M., Goldstine, J. V., & Gatti, R. A. (2007). ATM 缺陷淋巴母细胞线粒体内在功能障碍. Hum. Mol. Genet., 16(18), 2154 - 2164.
Amo, T., et al. (2011). PINK1 缺失引起的线粒体膜电位降低并非源于质子漏渗,而是呼吸链缺陷. Neurobiol. Dis., 41(1), 111 - 118.
Aras, S., et al. (2013). 细胞色素 c 氧化酶亚基 4 - 2 基因的氧依赖性表达受转录因子 RBPJ、CXXC5 和 CHCHD2 调控. Nucleic Acids Res., 41(4), 2255 - 2266.
Aras, S., et al. (2015). MNRR1 (原 CHCHD2) 是线粒体代谢的双细胞器调节因子. Mitochondrion, 20, 43 - 51.
Aras, S., et al. (2020). 线粒体核逆行调节因子 1(MNRR1)通过诱导稳态机制挽救 MELAS 细胞表型. Proc. Natl. Acad. Sci. USA, 117(50), 32056 - 32065.
Bai, P., et al. (2015). 聚 ADP - 核糖聚合酶作为线粒体活性调节因子. Trends Endocrinol. Metab., 26(2), 75 - 83.
Bakkenist, C. J., & Kastan, M. B. (2003). DNA 损伤通过分子间自磷酸化和二聚体解离激活 ATM. Nature, 421(6922), 499 - 506.
Beites, C. L., et al. (2005). 嗅觉上皮神经干细胞的鉴定与分子调控. Exp. Cell Res., 306(2), 309 - 316.
Chen, P., et al. (2003). 氧化应激导致共济失调 - 毛细血管扩张突变小鼠浦肯野神经元存活缺陷和树突发生障碍. J. Neurosci., 23(36), 11453 - 11460.
Chen, W. M., et al. (2023). DNA - PKcs 与 ATM 通过调节线粒体 ADP - ATP 交换作为氧化应激检查点机制. EMBO J., 42(6), e112094.
Cheng, A., et al. (2018). ATM 与 ATR 在兴奋性和抑制性囊泡群体行为中发挥互补作用. Proc. Natl. Acad. Sci. USA, 115(2), E292 - E301.
Chow, H. M., et al. (2019). ATM 因 ATP 耗竭被激活并通过 NRF1 调节线粒体功能. J. Cell Biol., 218(3), 909 - 928.
Cirot, C., et al. (2021). ATM 的氧化还原激活增强 GSNOR 翻译以维持线粒体自噬和氧化应激耐受性. EMBO Rep., 22(1), e50500.
Corti, A., et al. (2019). 共济失调 - 毛细血管扩张症患者 iPSC 来源神经元中的 DNA 损伤与转录调控. Sci. Rep., 9(1), 651.
Ditch, S., & Paull, T. T. (2012). ATM 蛋白激酶与细胞氧化还原信号:超越 DNA 损伤响应. Trends Biochem. Sci., 37(1), 15 - 22.
Fang, E. F., et al. (2014). XPA 通过 PARP - 1 过度活化和 NAD (+) 及 SIRT1 减少导致线粒体自噬缺陷. Cell, 157(4), 882 - 896.
Fang, E. F., et al. (2016). 补充 NAD (+) 通过线粒体自噬和 DNA 修复延长共济失调 - 毛细血管扩张症模型寿命并改善健康状态. Cell Metab., 24(4), 566 - 581.
Funayama, M., et al. (2015). 常染色体显性遗传晚发型帕金森病中的 CHCHD2 突变:全基因组连锁与测序研究. Lancet Neurol., 14(3), 274 - 282.
Guo, Z., et al. (2010). 氧化应激激活 ATM. Science, 330(6003), 517 - 521.
Hossain, M. B., et al. (2009). 聚 ADP - 核糖聚合酶 1 与核呼吸因子 1(NRF - 1)相互作用并参与 NRF - 1 转录调控. J. Biol. Chem., 284(13), 8621 - 8632.
Ikeda, A., et al. (2019). CHCHD2 突变导致 α - 突触核蛋白聚集. Hum. Mol. Genet., 28(23), 3895 - 3911.
Ikeda, A., Imai, Y., & Hattori, N. (2022). 神经退行性疾病相关线粒体蛋白 CHCHD2 与 CHCHD10—— 如何区分二者?Front. Cell Dev. Biol., 10, 996061.
Imai, Y., et al. (2019). 孪生 CHCH 蛋白 CHCHD2 与 CHCHD10:帕金森病、肌萎缩侧索硬化和额颞叶痴呆的关键分子. Int. J. Mol. Sci., 20(4).
Jansen, I. E., et al. (2015). CHCHD2 与帕金森病. Lancet Neurol., 14(7), 678 - 679.
Kamsler, A., et al. (2001). ATM 缺陷小鼠脑部氧化还原状态改变证实共济失调 - 毛细血管扩张症存在氧化应激增强. Cancer Res., 61(5), 1849 - 1854.
Kann, O., & Kovacs, R. (2007). 线粒体与神经元活动. Am. J. Phys. Cell Phys., 292(2), C641 - C657.
Kee, T. R., et al. (2021). 线粒体 CHCHD2:疾病相关突变、生理功能及当前动物模型. Front. Aging Neurosci., 13, 660843.
Kozlov, S. V., et al. (2016). 活性氧(ROS)激活的 ATM 依赖性磷酸化底物通过大规模磷酸化蛋白质组筛选鉴定. Mol. Cell Proteomics, 15(3), 1032 - 1047.
Lavin, M. F. (2013). 小鼠模型对共济失调毛细血管扩张症的适用性研究:存在神经功能缺陷但无神经退行性变. DNA 修复, 12(8), 612 - 619.
Lax, N. Z., et al. (2012). 线粒体 DNA 疾病患者的小脑性共济失调:一项分子临床病理学研究. 神经病理学与实验神经病学杂志, 71(2), 148 - 161.
Lee, J. H., & Paull, T. T. (2020). 线粒体在 ATM 介导的应激信号与活性氧调节中的核心作用. 氧化还原生物学, 32, 101511.
Lee, P., et al. (2013). SMRT 化合物可消除患者特异性 hiPSCs 神经衍生细胞中毛细血管扩张性共济失调的细胞表型. 自然通讯, 4, 1824.
Leeson, H. C., et al. (2021a). 通过患者嗅觉活检生成的共济失调毛细血管扩张症 iPSC 系鉴定出新的致病突变. 干细胞研究, 56, 102528.
Leeson, H. C., et al. (2021b). 利用对照组嗅觉黏膜活检重编程人嗅觉神经球衍生细胞. 干细胞研究, 56, 102527.
Li, J., et al. (2009). 神经元中胞质 ATM 蛋白调控突触功能. 当代生物学, 19(24), 2091 - 2096.
Liu, Y., & Zhang, Y. (2015). CHCHD2 连接线粒体代谢与细胞凋亡. 分子细胞肿瘤学, 2(4), e1004964.
Liu, Y., et al. (2015). CHCHD2 通过与 Bcl - xL 相互作用调控 Bax 激活来抑制细胞凋亡. 细胞死亡与分化, 22(6), 1035 - 1046.
Liu, W., et al. (2020). Chchd2 通过调节 Opa1 水平调控线粒体形态. 细胞死亡与分化, 27(6), 2014 - 2029.
Lopriore, P., et al. (2022). 线粒体共济失调:分子分型与临床异质性. 国际神经病学, 14(2), 337 - 356.
Luo, S., et al. (2023). ATM 通过调控 Akt/Drp1 介导的线粒体稳态保护脂多糖诱导的血脑屏障破坏. 休克, 60(1), 100 - 109.
Mackay - Sim, A. (2012). 综述:患者来源的嗅觉干细胞:脑疾病的新模型. 干细胞, 30(11), 2361 - 2365.
Madabhushi, R., et al. (2015). 活动诱导的 DNA 断裂调控神经元早期应答基因表达. 细胞, 161(7), 1592 - 1605.
Martini, H., & Passos, J. F. (2023). 细胞衰老:所有通路终归线粒体. FEBS 杂志, 290(5), 1186 - 1202.
Matsuoka, S., et al. (2007). ATM 与 ATR 底物分析揭示响应 DNA 损伤的广泛蛋白质网络. 科学, 316(5828), 1160 - 1166.
Miwa, S., et al. (2022). 细胞衰老与衰老过程中的线粒体功能障碍. 临床研究杂志, 132(13).
Mollo, N., et al. (2020). 唐氏综合征与衰老中线粒体网络结构的靶向研究. 国际分子科学杂志, 21(9).
Morita, A., et al. (2014). 线粒体是胞外氧化应激激活人肝癌细胞系 Hep G2 中 ATM 所必需的. 生物化学与生物物理研究通讯, 443(4), 1286 - 1290.
Navrkalova, V., et al. (2015). 氧化应激作为 ATM 缺陷型慢性淋巴细胞白血病患者的治疗视角. 血液学, 100(8), 994 - 996.
Nayler, S., et al. (2012). 共济失调毛细血管扩张症诱导多能干细胞重现细胞表型. 干细胞转化医学, 1(7), 523 - 535.
Nayler, S., et al. (2017). A - T 患者成纤维细胞的慢病毒重编程为诱导多能干细胞. 分子生物学方法, 1599, 401 - 418.
Ovchinnikov, D. A., et al. (2020). 纠正两名共济失调毛细血管扩张症患者 iPS 细胞中的 ATM 突变可恢复 DNA 损伤和氧化应激反应. 人类分子遗传学, 29(6), 990 - 1001.
Pizzamiglio, L., et al. (2016). ATM 在发育过程中调控 GABA 能张力的新功能. 大脑皮层, 26(10), 3879 - 3888.
Pizzamiglio, L., Focchi, E., & Antonucci, F. (2020). ATM 蛋白激酶在神经元通路与脑回路中的传统及新发现作用. 细胞, 9(9).
Purandare, N., et al. (2018). 细胞应激蛋白 CHCHD10 与 MNRR1 (CHCHD2):线粒体及核功能的协同作用与功能障碍. 生物化学杂志, 293(17), 6517 - 6529.
Quick, K. L., & Dugan, L. L. (2001). 超氧化物应激鉴定共济失调 - 毛细血管扩张症模型中濒危神经元. 神经病学年鉴, 49(5), 627 - 635.
Reichenbach, J., et al. (2002). 共济失调 - 毛细血管扩张症患者体内升高的氧化应激. 抗氧化剂与氧化还原信号, 4(3), 465 - 469.
Rimkus, S. A., et al. (2008). string/CDC25 突变抑制共济失调 - 毛细血管扩张症果蝇模型的细胞周期重启与神经退行性变. 基因与发育, 22(9), 1205 - 1220.
Salminen, A., Ojala, J., & Kaarniranta, K. (2011). 细胞凋亡与衰老:凋亡抗性增强会加速衰老过程. 细胞与分子生命科学, 68(6), 1021 - 1031.
Sarkar, A., et al. (2021). 共济失调 - 毛细血管扩张症突变蛋白与 Parkin 相互作用并诱导不依赖激酶活性的线粒体自噬 —— 来自套细胞淋巴瘤的证据. 血液学, 106(2), 495 - 512.
Scarpulla, R. C. (2008). 哺乳动物线粒体生物合成与功能的转录范式. 生理学评论, 88(2), 611 - 638.
Seager, R., et al. (2020). 神经元中线粒体定位与动态的机制及作用. 神经元信号, 4(2), NS20200008.
Sherman, B. T., et al. (2022). DAVID:用于基因列表功能富集分析及功能注释的网络服务器(2021 年更新). 核酸研究, 50(W1), W216 - W221.
Shi, Y., Kirwan, P., & Livesey, F. J. (2012). 人多能干细胞向大脑皮层神经元及神经网络的定向分化. 自然实验手册, 7(10), 1836 - 1846.
Shiloh, Y. (2020). 共济失调 - 毛细血管扩张症的小脑变性:基因组不稳定的典型案例. DNA 修复, 95, 102950.
Shiloh, Y., & Lederman, H. M. (2017). 共济失调 - 毛细血管扩张症(A-T):早衰的新维度. 衰老研究评论, 33, 76–88.
Shiloh, Y., & Ziv, Y. (2013). ATM 蛋白激酶:调控细胞对基因毒性应激的反应及其他. 自然评论分子细胞生物学, 14(4), 197–210.
Singh, C. K., et al. (2018). Sirtuins 在抗氧化和氧化还原信号中的作用. 抗氧化剂与氧化还原信号, 28(8), 643–661.
Stewart, R., et al. (2013). 用于共济失调 - 毛细血管扩张症的患者来源嗅干细胞疾病模型. 人类分子遗传学, 22(12), 2495–2509.
Stracker, T. H., et al. (2013). ATM 信号网络在发育与疾病中的作用. 遗传学前沿, 4, 37.
Valentin-Vega, Y. A., et al. (2012). 共济失调 - 毛细血管扩张症中的线粒体功能障碍. 血液, 119(6), 1490–1500.
Vizioli, M. G., et al. (2020). 线粒体至细胞核的逆行信号驱动衰老中胞质染色质形成及炎症. 基因与发育, 34(5–6), 428–445.
Wai, T., & Langer, T. (2016). 线粒体动力学与代谢调控. 内分泌与代谢趋势, 27(2), 105–117.
Watters, D., et al. (1999). 部分核外 ATM 定位于过氧化物酶体. 生物化学杂志, 274(48), 34277–34282.
Wiley, C. D., et al. (2016). 线粒体功能障碍诱导具有独特分泌表型的衰老. 细胞代谢, 23(2), 303–314.
Xu, J., et al. (2018). 大脑 SIRT1 介导代谢稳态与神经保护. 内分泌学前沿(洛桑), 9, 702.
Yang, B., et al. (2021). 补充 NAD + 通过改善线粒体自噬预防共济失调 - 毛细血管扩张症中 STING 诱导的衰老. 衰老细胞, 20(4), e13329.
Yeo, A. J., et al. (2021a). 共济失调 - 毛细血管扩张症中内质网 - 线粒体信号传导受损. 交叉科学, 24(1), 101972.
Yeo, A. J., et al. (2021b). 纠正共济失调 - 毛细血管扩张症(A-T)线粒体功能障碍的回补疗法. 分子代谢, 54, 101354.
Zhan, H., et al. (2010). 氧化应激诱导血管内皮细胞衰老中 ATM 介导的 DNA 损伤反应. 生物化学杂志, 285(38), 29662–29670.
Zhang, J., et al. (2015). ATM 在过氧化物酶体通过响应活性氧诱导 pexophagy. 自然细胞生物学, 17(10), 1259–1269.