¶ 自闭症相关SCN2A缺陷破坏人脑组装体中的皮质-纹状体回路
陈晓玲1,2,10、张景亮1,2,10、吴佳翔1,2、摩根·J·罗宾逊1,2,3、哈里什·科坦达拉曼4、刘艺恩1,2、伊里亚·M·冈萨雷斯·多佩索-雷耶斯5、托马斯·D·布菲努瓦5、马纳西·S·哈卢尔卡1,2、张在阳1,2、王慕寒1,2、艾琳·N·克里格6、赵元睿1,2、玛丽亚·I·奥利韦罗-阿科斯塔1,2、凯尔·W·韦茨丘拉克1,2、阙哲夫1,2、袁崇礼2,3、艾莉森·J·沙瑟2,7、纳迪亚·A·兰曼4,8、让-克里斯托夫·罗谢1,2、威廉·C·斯卡恩斯9、埃里克·J·克雷默5,11、杨阳 ^
1 Borch美国印第安纳州西拉法叶普渡大学药学院药物化学与分子药理学系,邮编 47907
2 美国印第安纳州西拉法叶普渡大学综合神经科学研究所,邮编 47907
3 美国印第安纳州西拉法叶普渡大学戴维森化学工程学院,邮编 47907
4 美国印第安纳州西拉法叶普渡大学癌症研究所,邮编 47907
5 法国蒙彼利埃蒙彼利埃大学及法国国家科学研究中心分子遗传学研究所
6 美国印第安纳州西拉法叶普渡大学理学院,邮编 47907
7 美国印第安纳州西拉法叶言语语言与听觉科学系,邮编 47907
8 美国印第安纳州西拉法叶普渡大学兽医学院比较病理生物学系,邮编 47907
9 美国康涅狄格州法明顿杰克逊基因组医学实验室,邮编 06032
10 这些作者贡献相同
11 资深作者
12 通讯联系人
*通讯邮箱:yangyang@purdue.edu
¶ 总结
重度自闭症谱系障碍(ASD)通常可归因于单基因突变,其中SCN2A(电压门控钠通道 )的蛋白质截短变异体(PTVs)是外显率最高的突变之一。尽管皮层-纹状体神经回路被认为是ASD的关键节点,但SCN2A缺失对人类神经回路的影响尚不清楚。通过人类皮层-纹状体组合体模型,我们发现导致自闭症的PTV SCN2A-C959X会损害长程皮层轴突投射、降低纹状体树突棘密度,并减弱兴奋性皮层-纹状体突触传递。出乎意料的是,这些携带SCN2A杂合无义突变的组合体表现出显著的网络过度兴奋性——这一人类细胞特异性表型在 小鼠中未被观察到,揭示了人类特异性神经回路的脆弱性。总之,我们的研究揭示了SCN2A缺失及SCN2A介导的ASD在人类特异性神经回路中的功能障碍。
¶ 关键词
自闭症, , SCN2A-C959X, 脑类器官, 组装体, 皮质-纹状体回路, 活细胞成像
标题:SCN2A-C959X突变损害皮质-纹状体回路功能
¶ 亮点
• 轴突投射促进人脑类组装体中的突触形成和功能连接。
• 在人类大脑类组装体中沿神经元轴突表达,并延伸至胞体和树突。
• SCN2A-C959X破坏轴突投射模式,损害兴奋性突触传递,降低树突棘密度,并导致神经元兴奋性升高。
¶ 图文摘要
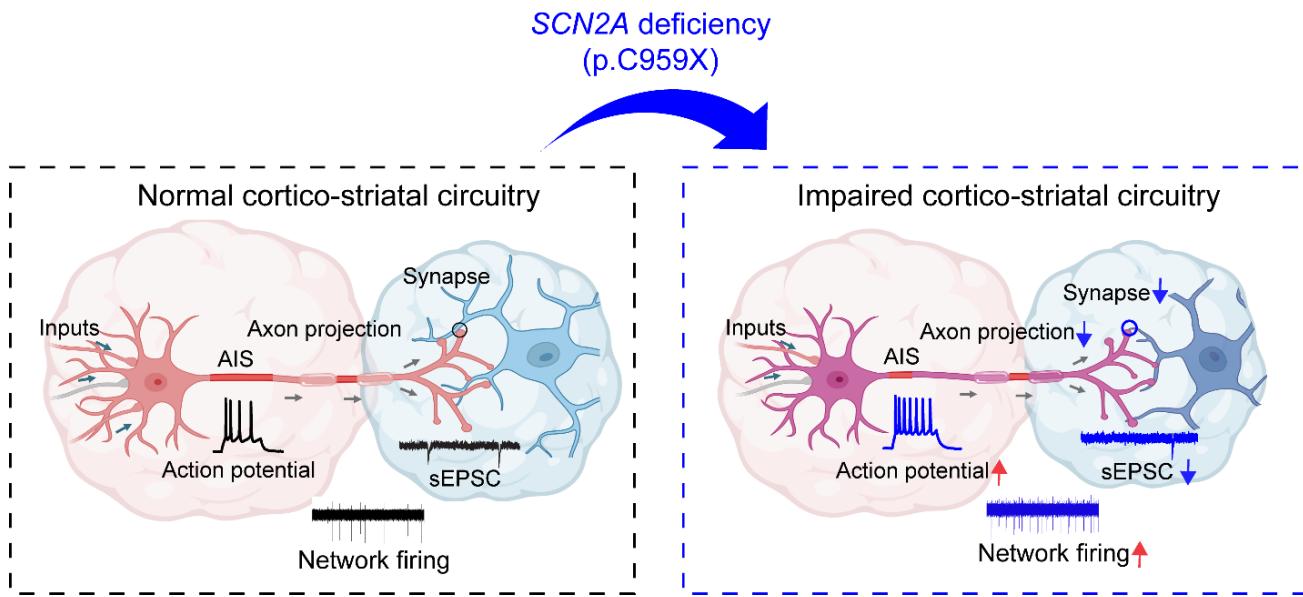
¶ 简而言之
¶ SCN2A单倍体功能不足损害皮质-纹状体神经回路。
SCN2A单倍体功能不足会破坏轴突起始段(AIS)的完整性,导致神经元过度兴奋(红色箭头)、轴突投射减少以及突触传递受损(sEPSCs降低和网络放电模式改变)。这些缺陷最终导致皮质-纹状体环路功能异常。
¶ 引言
自闭症谱系障碍(ASD)在美国约每31名儿童中即有一名患病1。尽管ASD在病因学上具有异质性2,但其严重表型常由单基因突变引起。其中,编码电压门控钠通道 的SCN2A基因功能缺失突变,已成为单基因型ASD的主要致病因素3,4。特别是新生无义突变SCN2A c.2877C>A(p.Cys959Ter),即SCN2A-C959X,会产生与严重临床表现相关的蛋白质截短变异体(PTV)3,5,6。 主要定位于轴突起始段,在发育中神经元的动作电位起始与传导过程中发挥关键作用。近期啮齿动物模型研究表明,还参与调控突触传递与树突功能7,8。然而,SCN2A缺陷如何干扰人脑神经元通讯机制仍不明确。随着ASD日益被确认为神经环路障碍(尤其涉及皮质-纹状体环路)9,10,阐明SCN2A缺陷如何影响该环路已成为关键课题。
新兴的诱导多能干细胞(iPSCs)来源的脑类器官已在体外11,12彻底改变了人类神经发育及神经发育障碍的研究。基于这些进展,整合区域特异性类器官的脑组装体13-18为跨脑区模拟神经元连接与环路功能提供了前所未有的机遇。本研究利用组装体技术重建了包含皮质锥体神经元与纹状体中型多棘神经元的皮质-纹状体神经网络。通过这一基于人类细胞的模型,我们系统性地探究了 缺失对人类神经元的影响。
本研究通过轴突投射的时空追踪发现,皮质轴突支配能够促进 assembloids 中纹状体神经元的突触形成与功能连接。相比之下,携带 SCN2A-C959X 突变的 assembloids 皮质投射减少,同时树突棘密度降低、兴奋性突触传递受损。RNA测序进一步证实了轴突及突触发育相关基因通路的表达下调。出乎意料的是,我们观察到这些 SCN2A缺陷神经元出现网络超兴奋现象,这可能源于受损突触功能的代偿性适应。总体而言,我们的研究结果揭示了 SCN2A缺陷在分子与环路层面的表现形式,证明了人脑 assembloids 在模拟自闭症谱系障碍相关环路功能障碍中的实用价值。这些发现深化了我们对 在重度自闭症谱系障碍相关神经环路中作用机制的理解。
¶ 结果
¶ 大脑中皮层投射促进纹状体突触形成与功能发育类器官组装体
为在体外模拟人类大脑皮层和纹状体的不同神经元类型及区域特性,我们依据已有方案13,利用人类诱导多能干细胞(hiPSCs)培育出皮层类器官(hCOs)和纹状体类器官(hStrOs)(图S1A)。hCOs的免疫染色结果验证了成熟神经元标志物神经元核抗原(NeuN)+和微管相关蛋白2(MAP2)+(图S1B),以及皮层特异性层状标志物T-box脑转录因子1(TBR1)+和COUP-TF相互作用蛋白2(CTIP2)+的表达,同时GABA能神经元标志物谷氨酸脱羧酶1(GAD67)+表达极少(图S1C),这证实了其皮层特性。类似地,hStrOs则表现出强烈的GAD67+和多巴胺-cAMP调节磷蛋白32(DARPP32)+(图S1D)作为纹状体中型多棘神经元(MSNs)13的标志物,提示其具有GABA能富集的纹状体特征。
为了重建皮质-纹状体神经环路,我们将两种区域特异性类器官融合形成hCO-hStrO组装体(图1A)。免疫染色证实了皮质区(NeuN+/TBR1+)与纹状体区(GABA+)的独立分区结构(图1B、C;S1E)。为检测皮质神经元是否与纹状体神经元建立突触连接,我们采用顺行病毒示踪技术:将hCOs与AAV1-hSyn1-Cre共培养,同时将hStrOs分别与AAV1-Ef1a-DIO-mScarlet和AAV9-hSyn-EGFP共培养。若存在突触连接,AAV1-hSyn1-Cre将顺行转运至hStrO神经元并表达Cre重组酶,随后Cre活性将翻转DIO-mScarlet盒并诱导mScarlet表达,而EGFP+细胞则作为纹状体区标记物(图1D)。在融合形成组装体一个月后,我们在hStrOs区域观察到共表达GFP与mScarlet的细胞(图1D)。这些数据表明组装体内存在皮质-纹状体神经连接,此结果通过逆行标记实验得到进一步验证(图1E)。为评估皮质-纹状体连接的功能性,我们结合化学遗传学与膜片钳记录技术进行检测。为诱发突触前神经递质释放,用AAV9-hSyn-Gq-mCherry转导hCOs以表达Gq-DREADD(经CNO给药可激活神经元),同时用AAV9-hSyn-EGFP转导hStrOs进行分区标记。融合两个月后,脑片膜片钳记录显示:在CNO浴液给药后,hStrO神经元的自发兴奋性突触后电流(sEPSC)频率出现可逆性增加,这表明皮质与纹状体神经元间存在功能性突触连接(图1F(i–iv))。
已知皮层谷氨酸能投射对纹状体MSN的成熟至关重要19。为探究皮层投射如何影响纹状体突触特性,我们比较了皮质-纹状体装配体与非融合hStrO中纹状体神经元的sEPSC频率(图1G(i))。融合后两个月,我们观察到在相同体外发育阶段,hCO-hStrO中的sEPSC频率高于单独的hStrO(图1G(ii–iv)),表明与hCO的连接增强了纹状体突触形成。进一步地,为确定突触形成增加是否与皮质-纹状体连接相关,我们通过双重病毒标记在装配体中区分了接收直接皮层输入的纹状体神经元(“接收神经元”)与未接收皮层输入的神经元(“非接收神经元”),并检测了它们的兴奋性输入。具体而言,hCO用AAV1-hSyn1-Cre标记,hStrO用AAV1-Ef1a-DIO-mScarlet和AAV9-hSyn-EGFP标记(图1H(i))。hStrO中的接收神经元(EGFP+/mScarlet+)比非接收神经元(仅EGFP+)表现出显著更高的树突棘密度及更高比例的成熟树突棘类型(图1H(ii–v))。这些结果共同表明,源自hiPSCs的皮质-纹状体装配体成功建立了促进神经元成熟的功能性连接,为研究神经发育障碍中的环路水平相互作用提供了稳健的体外模型。
¶ SCN2A-C959X突变破坏大脑组装体中的皮质-纹状体回路
Scn2a在皮质-纹状体神经回路20中持续稳定表达,该回路与自闭症谱系障碍(ASD)的病理机制密切相关{v11。为研究ASD相关的SCN2A缺陷如何影响该回路的神经元功能,我们利用CRISPR/Cas9基因组编辑技术,在参照系KOLF2.1J诱导多能干细胞(iPSCs)中引入SCN2A c.2877C>A(p.Cys959Ter)突变(该突变在重度自闭症患儿中发现)(图2A;S2A)。Sanger测序证实成功构建了杂合(HET)与纯合(HOM)突变细胞系以及同基因野生型(WT)对照组(图2A)。蛋白质印迹分析显示,与WT相比,HET和HOM人皮质类器官(hCOs)中Na 1.2蛋白水平显著降低(图2B)。定量PCR分析进一步表明其呈现剂量依赖性在 hCO-hStrO 组装体中出现 SCN2A mRNA 水平降低(图 2C),与该突变的 LoF/缺陷表型一致。利用小鼠脑组织进行的免疫组织化学检测将 信号定位于轴突起始段(AIS),即动作电位(AP)的起始位置7。我们的免疫染色显示 与 AIS 标志物 Ankyrin-G 共定位(图 2D),表明 通道在 hCO 和 神经元中的定位与小鼠脑研究结果一致。为探究 SCN2A 缺陷如何影响 AIS,我们对不同基因型的 AIS 长度进行了定量分析。有趣的是,我们发现 HET SCN2A-C959X神经元在 hCO 切片中显示出较短的 AIS 长度(图 S2B(i, ii)),提示 SCN2A 缺陷会损害 AIS 可塑性——该特性对神经元内在兴奋性具有关键影响21。出乎意料的是,全细胞膜片钳记录显示 SCN2A缺陷神经元相较于 WT 对照组表现出兴奋性增强,其特征表现为注入电流与 AP 放电的输入-输出关系曲线发生偏移(图 2E(i, ii))。这种神经元过度兴奋性进一步表现为:最大放电频率升高、输入电阻增加以及触发 AP 所需的电流阈值降低(图 2E(iii–v))。
为了评估SCN2A缺陷对超出神经元内在特性的脑环路的影响,我们检测了皮层-纹状体组装体的连接性。使用野生型类器官制备的组装体中,我们观察到融合后从人皮层类器官向人纹状体类器官的mScarlet+投射逐步增加(图2F(i, ii))。相比之下,在第20天、30天时,与野生型相比,HETSCN2A-C959X组装体向人纹状体类器官的轴突支配显著减少。以及融合后的42(图2F(iii, iv)),表明SCN2A缺失会损害长程脑区间投射。考虑到皮质投射受损对神经元成熟的潜在影响,我们进一步通过双重AAV标记分析了由皮质输入支配的hStrO接收神经元的形态。分别用AAV1-hSyn1-Cre或AAV1-Ef1a-DIO-mScarlet转导hCOs和hStrOs。融合一个月后,对hStrO区室中HET接收神经元(EGFP+/mScarlet{v7)的Sholl分析显示,其树突复杂度较野生型显著降低,包括树突分支减少、树突长度缩短以及交互点减少(图2G(i–v))。这些发现将表达下降与类组装体内轴突支配及神经元成熟受损联系起来。
先前在小鼠中的研究结果也表明 定位于胞体和树突8。与此一致,我们发现在人类组装体的皮层和纹状体区域中, 与神经元胞体标记物(NeuN+)及神经突标记物(MAP2+)共定位(图2H、S2D),提示 在胞体和树突中发挥作用。为评估SCN2A-C959X对兴奋性突触的影响,我们首先通过Syn1标记突触前末梢、PSD95标记突触后位点,确认皮层轴突能够形成兴奋性突触。与对照组相比,对Syn1+/PSD95+免疫反应性的定量分析显示,在HET SCN2A-C959X神经元中两者的共定位显著减少,表明兴奋性突触连接受损(图S2C(i–iii))。鉴于树突棘是兴奋性突触的主要位点,我们进一步在活体hCO-hStrO组装体中表征了树突棘形态。与野生型相比,HET接收神经元(mScarlet+)的树突棘密度显著降低,其中细长型、粗短型及成熟蘑菇型树突棘受影响尤为明显(图2I(i–iii)),提示树突棘存在结构缺陷。为探究这些形态学变化是否导致功能损伤,我们在hCO-hStrO组装体切片中对接收神经元(mScarlet+)进行膜片钳记录(图2J(i))。HET接收神经元自发性兴奋性突触后电流频率较野生型显著降低(图2J(ii–iv)),表明突触传递功能受损。对记录神经元的免疫染色进一步证实其树突棘密度较低(图S2E(i–v)),与活细胞成像结果一致(图2I(i–iii))。
为进一步增强研究结果的严谨性,我们还检测了携带SCN2A-C959X纯合突变的组装体,并观察到相似的表型,包括树突复杂性降低和树突棘密度减少(数据未展示)。总体而言,我们的数据表明SCN2A-C959X突变会破坏人类皮质-纹状体组装体的轴突起始段可塑性、轴突投射、树突形态结构及突触功能。这些综合表型凸显了 在建立和维持皮质-纹状体环路功能连接中的关键作用,为理解SCN2A功能缺失变异如何导致自闭症谱系障碍的病理机制提供了新的视角。
SCN2A-C959X突变损害轴突和突触相关基因通路为阐明SCN2A缺陷在皮质-纹状体回路中的分子基础,我们对不同基因型的hCO-hStrO组装体进行了批量RNA测序(图3A)。主成分分析(PCA)显示WT、HET与HOM组间存在明显聚类,揭示了基因表达谱的整体性变化(图S3A)。我们验证了SCN2A表达水平——在HET和HOM组中均显著降低(图3B),与图2C所示的qPCR结果一致。随后我们在HET vs. WT比较中鉴定出6,145个差异表达基因(2,846个上调;3,299个下调;图3C(i)),在HOM vs. WT比较中鉴定出8,907个差异表达基因(4,717个上调;4,190个下调;图3C(ii))。后续分析优先聚焦于HET与HOM组共有的重叠差异表达基因,因为这些基因可能代表SCN2A缺陷的核心致病机制。鉴于我们发现SCN2A-C959X会影响神经元兴奋性与突触传递(图2E、J),我们进一步探索了与这些功能相关的基因。分析结果显示,电压门控钠通道(SCN2A、SCN8A、SCN1A)、轴突完整性(ANK2、ANK3)及突触通路(SYN1、DLG4、GRIA2、GRIA3、GRIA4、GRIN1、GRIN2A、GRIN3A)等关键基因均出现显著下调(图3C(i, ii))。
对重叠差异表达基因(DEGs)的进一步研究发现,有2,286个基因持续下调(图3D(i))。对这些基因的KEGG通路富集分析显示,与神经发育、轴突导向和突触信号传导相关的通路存在显著紊乱(图3D(ii))。GO细胞组分(CC)富集分析进一步强化了轴突末梢、轴突起始段(AIS)、兴奋性突触和GABA能突触存在缺陷的结论(图3D(iii, iv);S3B)。观察到的ANK3(锚蛋白-G)和DLG4(PSD-95)下调与我们免疫染色实验中的蛋白质水平变化一致(图S2B, C)。此外,GO分子功能(MF)和Reactome分析证实了SCN2A缺陷对突触通路的主要影响(图S3C, E)。另外,GO生物过程(BP)分析突出显示了多个离子通道类别(特别是配体门控通道以及电压门控钠离子和钾离子通道)的显著下调(图S3D(i–iv))。这种失调可能解释了在SCN2A-C959X突变神经元中观察到的神经元兴奋性改变。
相反,通过对重叠上调基因(2,052个基因;图3E(i))进行Reactome通路分析发现,这些基因在无义介导的mRNA降解(NMD)、mRNA加工、mRNA剪接以及外显子连接复合体(EJC)活性等过程中显著富集(图3E(ii, iii);S3F),这表明RNA监视机制得到了增强。选择Reactome是因为其对RNA代谢通路具有全面的注释,而其他数据库(如KEGG)对这些通路的覆盖相对有限。鉴于我们的蛋白质印迹和qPCR结果显示SCN2A mRNA和蛋白水平大幅下降(图2B、C),这些分子谱分析结果表明C959X突变很可能通过激活NMD相关通路导致SCN2A表达降低。
有趣的是,网络分析揭示了59个自闭症相关差异表达基因在HET和HOM类组装体中均发生一致性改变(图 S3G)。排名前10的自闭症相关基因热图(图3F)展示了它们的共表达模式,为不同遗传风险因素可能如何共同驱动ASD病理22提供了共享神经生物学机制的新见解。总体而言,我们的转录组分析表明,SCN2A-C959X显著破坏轴突与突触通路,损害人类皮质-纹状体回路中的突触连接与神经元功能。这种调控异常可能是ASD相关神经功能障碍的基础,从而为SCN2A功能缺失变异影响ASD病因学提供了分子层面的解释。
¶ 讨论
自闭症谱系障碍是一种复杂的神经发育状况,而识别由单基因突变(如SCN2A基因突变)引起的分子扰动,能为病理生理学提供重要见解。本研究利用人皮层-纹状体组装体模型,探究SCN2A-C959X突变如何破坏神经元功能与环路连接性。我们的研究结果表明,SCN2A缺陷会导致轴突投射、树突棘形态、突触传递和神经元兴奋性出现显著障碍,这突显了在建立和维持功能性皮层-纹状体环路中的关键作用。
在神经元中扮演多重角色:它从轴突起始段(AIS)延伸至中段和远端轴突,决定动作电位(AP)的起始与传播,同时亦定位于胞体和树突,调控AP的反向传播7,8。与之相符,我们的免疫染色结果显示,在人源组装体中, 与锚蛋白G共定位于AIS,与NeuN共定位于胞体,与MAP2共定位于树突(图2)。在病理条件下,轴突中SCN2A的缺失可能减弱AP传播过程中的峰值,从而可能导致神经递质释放减少、轴突投射及突触连接受损。我们观察到SCN2A-C959X突变破坏了皮层轴突向纹状体的投射,导致纹状体接收端中型多棘神经元的树突棘密度降低、形态改变以及突触传递功能受损(图2)。这些发现与啮齿类动物研究结果一致,均表明 功能障碍会损害AP反向传播,最终导致突触功能缺陷7,8。此外,我们发现SCN2A-C959X会导致AIS缩短——这一发现尤为值得关注,因为通常更长的AIS长度与神经元内在兴奋性增强相关,这是由于更长的AIS中Na 电导增加会降低AP电压阈值21。矛盾的是,尽管AIS缩短,我们模型中的SCN2A缺陷皮层神经元却表现出内在兴奋性亢进(图2)。有趣的是,这种反直觉的兴奋性亢进与钾通道的下调同时发生(图S3),这竟与我们之前在Scn2A缺陷小鼠中的观察结果惊人地一致20。我们提出,钾通道下调可能是抵消缺失的代偿机制,最终导致神经元兴奋性过度20。
值得注意的是,杂合型 SCN2A 无义突变(会使 SCN2A 表达量降低约 50%)引发的神经元过度兴奋,是一种人类细胞特异性表型 —— 小鼠模型需将 Scn2a 表达量降低 70% 以上才会表现出类似的过度兴奋状态20,23,这既凸显了人类与小鼠细胞生理学之间的关键差异,也强调了利用人类细胞模型研究人类疾病的必要性。类器官与组装体模型的另一优势在于,它们能在较长周期(数百天)内持续成熟,使神经元可以发育出复杂的结构 ²⁴和电生理特性 ²⁵,与生理状态高度相似 ²⁶;这些先进特性或许可以解释我们的研究结果,与此前使用 2D 培养的 SCN2A⁺/⁻人诱导多能干细胞衍生神经元的研究之间存在的差异,具体而言,2D培养与我们在本研究中的发现不同(图2),SCN2A+/-神经元表现出轴突起始段长度增加和神经元兴奋性降低27。已知在二维单层培养的神经元处于相对未成熟阶段28。相比之下,三维组装体的增强成熟度可能揭示在早期发育阶段无法检测到的表型。
皮层-纹状体环路中的主要神经元通常不产生自发放电(图2E(i, ii);4E(i, ii)),但SCN2A-C959X神经元具有内在超兴奋性(图2)。尽管存在兴奋性传递受损,MEA记录显示SCN2A-C959X组装体神经元放电频率增加(图4)。这种显著的可逆性网络水平超兴奋性可能源于代偿性突触变化(例如兴奋性与抑制性突触的可能失衡;图3;S3),与我们先前在Scn2a缺陷小鼠中的发现一致—该模型同时表现出突触功能障碍和体内神经元放电增强29。这引发了两个引人深思的问题:1)相较于单纯的内在兴奋性,包括轴突投射、树突棘密度和突触神经传递在内的环路水平缺陷是否与ASD发病机制更密切相关?2)神经元通信的长期损伤是否会触发代偿性内在超兴奋性以抵消神经递质释放的减少?未来研究中解答这些问题将进一步阐明疾病病理中神经元兴奋性调控的复杂性。
SCN2A基因突变在患者中呈杂合状态。SCN2A-C959X无义突变导致提前出现终止密码子,可能引发无义介导的mRNA降解6,30,进而产生单倍剂量不足。在此情况下,未受影响等位基因的50%表达量(图2)不足以维持正常神经元功能。我们的RNA-seq数据显示NMD通路上调(图3),进一步支持单倍剂量不足是主要致病机制。相比之下,SCN2A功能缺失型突变(尤其是错义突变)也可能导致显性负效应31-33,产生的突变蛋白会聚集并扣押野生型 ,进一步降低功能性水平。虽然我们的纯合型SCN2A-C959X模型未模拟患者体内的杂合状态,但它代表了SCN2A缺陷的极端形式,可能揭示人类神经元中SCN2A功能相关的重要生物学机制。综上,我们研究结果表明同时携带杂合与纯合SCN2A突变的人类大脑组装体可作为研究自闭症谱系障碍细胞与环路病理学的强大平台。
总之,我们的研究表明,导致自闭症的SCN2A-C959X突变会严重破坏人脑组装体的皮质-纹状体连接,进而引发突触缺陷、基因表达改变和网络高兴奋性。从概念上讲,这项工作通过直接将物种特异性神经环路破坏与人类病理相关的功能结果联系起来,显著超越了先前Scn2a小鼠模型的研究。更广泛而言,这些发现凸显了人脑类器官/组装体在模拟神经发育障碍中的强大能力,以及推进精准医疗方法的潜力,为适用于其他单基因自闭症谱系障碍的研究奠定了基础框架。
资源可用性主要联系人
更多信息或获取资源的请求,请联系主要负责人杨洋(yangyang@purdue.edu),相关事宜将由她处理。
¶ 材料可用性
本研究未生成新的独特试剂。
数据和代码可用性
支撑本研究的数据集和源代码详情,可根据要求向通讯作者索取。
¶ 致谢
本研究报道的研究工作部分获得了美国国立卫生研究院国家神经疾病与中风研究所的资助(项目号R01NS117585和R01NS123154,资助对象:Y.Y.)。作者诚挚感谢普渡大学药物发现研究所和普渡大学整合神经科学研究所提供的额外资金支持。X.C. 获得了美国癫痫学会博士后研究奖学金的支持。杨实验室感谢FamilieSCN2A基金会授予Y.Y.的霍奇金-赫胥黎研究奖,以及授予X.C.、J.Z.和Y.E.Y.的动作电位研究资助。杨实验室感谢癌症生物信息学协作核心( )提供的生物信息学支持,该核心由印第安纳大学西蒙综合癌症中心(资助号P30CA082709)、普渡大学癌症研究所(资助号P30CA023168)和沃尔瑟癌症基金会共同支持。本文内容仅代表作者责任,不一定反映印第安纳州卫生部门或美国国立卫生研究院的官方观点。
作者贡献
X.C.、J.Z.和Y.Y.设计了实验。X.C.、J.Z.、M.J.R.、M.S.H.、Z.Z.、M.W.和E.N.C.参与了相关工作。进行了实验。W.C.S.设计并进行了SCN2A基因编辑实验。I.G.D.R.、T.B.和E.J.K.制备并提供了未公开的实验试剂。J.W.、H.K.、Y.-E.Y.、Y.Z.、M.I.O.A.、K.W.W.、Z.Q.、C.Y.、A.J.S.、N.A.L.、J.-C.R.和W.C.S.参与了数据分析和实验设计。Y.Y.负责监督该项目。X.C.、J.Z.和Y.Y.在全体作者的共同参与下撰写了论文。
利益冲突声明作者声明不存在利益冲突。
生成式人工智能和人工智能辅助技术在写作过程中的使用声明在准备本作品的过程中,作者使用ChatGPT 4.5来提升文稿的可读性与语言质量,同时确保主要结论保持不变。使用该工具后,作者对文字进行了必要的审阅与编辑,并对出版物内容承担全部责任。
¶ 参考文献
- Shaw, K.A. (2025). 4岁和8岁儿童孤独症谱系障碍的患病率与早期识别——美国16个监测点,2022年孤独症与发育障碍监测网络。MMWR. 监测摘要 74。
- Klei, L., Sanders, S.J., Murtha, M.T., Hus,V., Lowe, J.K., Willsey, A.J., Moreno-De-Luca, D., Yu, T.W., Fombonne, E., Geschwind, D., 等. (2012).常见的遗传变异以加性方式作用是孤独症的主要风险来源。Mol Autism 3, 9. 10.1186/2040-2392-3-9.
- Sanders, S.J., Murtha, M.T., Gupta, A.R., Murdoch, J.D., Raubeson, M.J., Willsey, A.J., Ercan-Sencicek, A.G., DiLullo, N.M., Parikshak, N.N., Stein, J.L., 等. (2012). 全外显子组测序揭示的新生突变与孤独症密切相关。Nature 485, 237–241. 10.1038/nature10945.
- Satterstrom, F.K., Kosmicki, J.A., Wang, J.,Breen, M.S., De Rubeis, S., An, J.Y., Peng, M., Collins, R., Grove, J., Klei, L., 等. (2020). 大规模外显子组测序研究揭示了孤独症神经生物学中发育性和功能性的变化。Cell 180, 568–584.e523. 10.1016/j.cell.2019.12.036.
- Uddin, M., Tammimies, K., Pellecchia, G., Alipanahi, B., Hu, P., Wang, Z., Pinto, D.,Lau, L., Nalpathamkalam, T., Marshall, C.R., 等. (2014). 受纯化选择的大脑表达外显子富含孤独症谱系障碍的新生突变。Nat Genet 46, 742–747. 10.1038/ng.2980.
- Ben-Shalom, R., Keeshen, C.M., Berrios, K.N., An, J.Y., Sanders, S.J., 和 Bender, K.J. (2017). 对Na(V)1.2功能的相反效应是SCN2A变异在孤独症谱系障碍个体或婴儿癫痫患者中观察到的差异的基础。Biol Psychiatry 82, 224–232. 10.1016/j.biopsych.2017.01.009.
- Spratt, P.W.E., Ben-Shalom, R., Keeshen, C.M., Burke, K.J., Jr., Clarkson, R.L., Sanders, S.J., 和 Bender, K.J. (2019). 孤独症相关基因Scn2a促进前额叶皮层树突兴奋性与突触功能。Neuron 103, 673–685 e675. 10.1016/j.neuron.2019.05.037.
- Nelson, A.D., Catalfio, A.M., Gupta, J.P., Min, L., Caballero-Floran, R.N., Dean, K.P., Elvira, C.C., Derderian, K.D., Kyoung, H., Sahagun, A., 等. (2024). 孤独症风险基因Scn2a和Ank2在新皮层锥体细胞树突中的物理和功能汇聚。Neuron 112, 1133–1149 e1136. 10.1016/j.neuron.2024.01.003.
- Murugan, M., Jang, H.J., Park, M., Miller, E.M., Cox, J., Taliaferro, J.P., Parker, N.F., Bhave, V., Hur, H., Liang, Y., 等. (2017). 前额叶皮层下行投射中社会性与空间性编码的结合。Cell 171, 1663–1677 e1616. 10.1016/j.cell.2017.11.002.
- Ma, Z.-H., Lu, B., Li, X.,Mei, T., Guo, Y.-Q., Yang, L., Wang, H., Tang, X.-Z., Ji, Z.-Z., Liu, J.-R., 等. (2022). 孤独症谱系障碍中皮质-纹状体功能连接发育轨迹的非典型性。Autism 26, 1108–1122. 10.1177/13623613211041904.
- Jourdon, A., Wu, F., Mariani, J., Capauto, D., Norton, S., Tomasini, L., Amiri, A., Suvakov, M., Schreiner,J.D., Jang, Y., 等. (2023). 在前脑类器官中模拟特发性孤独症揭示了早期神经发生过程中兴奋性皮层神经元亚型的不平衡。Nat Neurosci 26, 1505–1515. 10.1038/s41593-023-01399-0.
- Huang, W.K., Wong, S.Z.H., Pather, S.R., Nguyen, P.T.T., Zhang, F., Zhang, D.Y., Zhang, Z., Lu, L., Fang, W., Chen, L.,等. (2021). 从人诱导多能干细胞生成下丘脑弓状核类器官。Cell Stem Cell 28, 1657–1670 e1610. 10.1016/j.stem.2021.04.006.
- Miura, Y., Li, M.Y., Birey, F., Ikeda, K., Revah, O., Thete, M.V., Park, J.Y., Puno, A., Lee, S.H., Porteus, M.H., 和 Pasca, S.P. (2020). 人纹状体类器官的生成和来自人类多能干细胞的皮质-纹状体组装体。《自然-生物技术》38, 1421–1430. 10.1038/s41587-020-00763-w.
- Andersen, J., Revah, O., Miura, Y., Thom, N., Amin, N.D., Kelley, K.W., Singh,M., Chen, X., Thete, M.V., Walczak, E.M., 等人. (2020). 功能性人类3D皮质-运动组装体的生成。《细胞》183, 1913–1929 e1926. 10.1016/j.cell.2020.11.017.
- Miura, Y., Li, M.-Y., Revah, O., Yoon, S.-J.,Narazaki, G., 和 Pasca, S.P. (2022). 构建大脑组装体以探究人类神经回路。《自然实验手册》17, 15–35. 10.1038/s41596-021-00632-z.
- Birey, F., Li, M.Y., Gordon, A., Thete, M.V., Valencia, A.M., Revah, O., Pasca, A.M., Geschwind, D.H., 和 Pasca, S.P. (2022). 在蒂莫西综合征前脑组装体中剖析人类中间神经元迁移的分子基础。《细胞干细胞》29, 248–264 e247. 10.1016/j.stem.2021.11.011.
- Kim, J.I., Miura, Y., Li, M.Y., Revah, O., Selvaraj, S., Birey, F., Meng, X., Thete, M.V., Pavlov, S.D., Andersen,J., 等人. (2024). 人类组装体揭示CACNA1G基因变异在丘脑皮质通路中的后果。《神经元》112, 4048–4059 e4047. 10.1016/j.neuron.2024.09.020.
- Onesto, M.M., Kim, J.I., 和 Pasca, S.P. (2024). 用于研究组织和疾病生物学的细胞-细胞相互作用组装体模型。《细胞干细胞》31, 1563–1573. 10.1016/j.stem.2024.09.017.
- Steiner, H., 和 Tseng, K.Y. (2016). 基底神经节结构与功能手册(学术出版社)。
- Zhang, J., Chen, X., Eaton, M., Wu, J., Ma, Z., Lai, S., Park, A., Ahmad, T.S., Que, Z., Lee, J.H., 等人. (2021). 电压门控钠通道Na(V)1.2的严重缺陷会提高成年小鼠的神经元兴奋性。《细胞报告》36. 10.1016/j.celrep.2021.109495.
- Harley, P., Kerins, C., Gatt, A., Neves, G., Riccio, F., Machado, C.B., Cheesbrough, A., R’Bibo, L., Burrone, J., 和 Lieberam, I. (2023). ALS患者hiPSC运动神经元的异常轴突起始段可塑性和内在兴奋性。《细胞报告》42. 10.1016/j.celrep.2023.113509.
- Paulsen, B., Velasco,S., Kedaigle, A.J., Pigoni, M., Quadrato, G., Deo, A.J., Adiconis, X., Uzquiano, A., Sartore, R., Yang, S.M., 等人. (2022). 自闭症基因汇聚于共享神经元类别的异步发育。《自然》602, 268–273. 10.1038/s41586-021-04358-6.
- Spratt, P.W.E., Alexander, R.P.D., Ben-Shalom, R., Sahagun, A., Kyoung, H., Keeshen, C.M., Sanders, S.J., 和 Bender, K.J. (2021). 新皮质锥体细胞中Na(V)1.2钠通道缺失引起的矛盾性过度兴奋。《细胞报告》36, 109483. 10.1016/j.celrep.2021.109483.
- Quadrato, G., Nguyen, T., Macosko, E.Z., Sherwood, J.L., Min Yang, S., Berger, D.R., Maria, N., Scholvin, J., Goldman, M., Kinney, J.P.,等人. (2017). 光敏感人脑类器官中的细胞多样性和网络动态。《自然》545, 48–53. 10.1038/nature22047.
- Fair, S.R., Julian, D., Hartlaub, A.M., Pusuluri, S.T., Malik, G., Summerfied, T.L., Zhao, G., Hester, A.B., Ackerman, W.E., Hollingsworth, E.W., 等人. (2020). 大脑类器官的电生理成熟与动态形态和细胞发育相关。《干细胞报告》15, 855–868. https://doi.org/10.1016/j.stemcr.2020.08.017.
- Pasca, A.M., Sloan, S.A., Clarke, L.E., Tian, Y., Makinson, C.D., Huber, N., Kim, C.H., Park, J.-Y., O’Rourke, N.A., Nguyen, K.D., 等人. (2015). 来自人类多能干细胞的3D培养功能性皮质神经元和星形胶质细胞。《自然方法》12, 671–678.
- Tamura, S., Nelson, A.D., Spratt, P.W.E., Kyoung, H., Zhou, X., Li, Z., Zhao, J., Holden, S.S., Sahagun, A., Keeshen, C.M., 等人 (2022). CRISPR激活挽救与SCN2A单倍体不足相关的自闭症谱系障碍异常。bioRxiv, 2022.2003.2030.486483. 10.1101/2022.03.30.486483.
- Nunes, C., Gorczyca, G., Mendoza-deGyves, E., Ponti, J., Bogni, A., Carpi, D., Bal-Price, A., 及 Pistollato, F. (2022). 提升生物复杂性以促进神经元与少突胶质细胞成熟并改善体外发育神经毒性(DNT)评估。Reproductive Toxicology 110, 124–140. https://doi.org/10.1016/j.reprotox.2022.03.017.
- Zhang, J., Eaton, M., Chen, X., Zhao, Y., Kant, S., Deming, B.A., Harish, K., Nguyen, H.P., Shu, Y., Lai, S., 等人 (2025). 恢复兴奋/抑制平衡可增强神经元信噪比并挽救自闭症相关Scn2a缺陷的社交障碍。bioRxiv, 2025.2003.2004.641498. 10.1101/2025.03.04.641498.
- Sanders, S.J., Campbell, A.J., Cottrell, J.R., Moller, R.S., Wagner, F.F., Auldridge, A.L., Bernier, R.A., Catterall, W.A., Chung, W.K., Empfield, J.R., 等人 (2018). SCN2A介导疾病的理解与治疗进展。Trends in neurosciences 41, 442–456. 10.1016/j.tins.2018.03.011.
- Epifanio, R., Giorda, R., Merlano, M.C., Zanotta, N., Romaniello, R., Marelli, S., Russo, S., Cogliati, F., Bassi, M.T., 及 Zucca, C. (2021). SCN2A致病性变异与癫痫:异质性的临床、遗传及诊断特征。Brain Sci 12. 10.3390/brainsci12010018.
- Carvill, G.L., Matheny, T., Hesselberth, J., 及 Demarest, S. (2021). 癫痫中的单倍体不足、显性负性与功能获得机制:使治疗方法与病理生理学相匹配。Neurotherapeutics 18, 1500–1514. 10.1007/s13311-021-01137-z.
- Gao, Y., Shonai, D., Trn, M., Zhao, J., Soderblom, E.J., Garcia-Moreno, S.A., Gersbach, C.A., Wetsel, W.C., Dawson, G., Velmeshev, D., 等人 (2024). 天然蛋白质组的邻近分析揭示自闭症及相关神经发育疾病小鼠模型的表型修饰因子。Nature communications 15, 6801. 10.1038/s41467-024-51037-x.
- Skarnes, W.C., Pellegrino, E., 及 McDonough, J.A. (2019). 提高人类干细胞中同源定向修复效率。Methods 164-165, 18–28. https://doi.org/10.1016/j.ymeth.2019.06.016.
- Skarnes, W.C., Ning, G., Giansiracusa, S., Cruz, A.S., Blauwendraat, C., Saavedra, B., Holden, K., Cookson, M.R., Ward, M.E., 及 McDonough, J.A. (2021). 利用dCas9控制人类干细胞中的同源定向修复结果。bioRxiv, 2021.2012.2016.472942. 10.1101/2021.12.16.472942.
- Pantazis, C.B., Yang, A., Lara, E., McDonough, J.A., Blauwendraat, C., Peng, L., Oguro, H., Kanaujiya, J., Zou, J., Sebesta, D., 等人 (2022). 用于大规模合作研究的人类诱导多能干细胞参考系。Cell Stem Cell 29, 1685–1702 e1622. 10.1016/j.stem.2022.11.004.
- Pasca, S.P., Arlotta, P., Bateup, H.S., Camp, J.G., Cappello, S., Gage, F.H., Knoblich, J.A., Kriegstein, A.R., Lancaster, M.A., Ming, G.L., 等人 (2025). 神经类器官、组装体与移植研究框架。Nature 639, 315–320. 10.1038/s41586-024-08487-6.
- Zhang, J., Zhang, C., Chen, X., Wang, B., Ma, W., Yang, Y., Zheng, R., 及 Huang, Z. (2021). PKA-RII自身磷酸化调控小鼠PKA活性与癫痫表型。Communications Biology 4, 263. 10.1038/s42003-021-01748-4.
- Wang, J., He, X., Meng, H., Li, Y., Dmitriev, P., Tian, F., Page, J.C., Lu, Q.R., 及 He, Z. (2020). 联合操控GPR17与小胶质细胞诱导再生轴突的稳健髓鞘形成。Neuron 108, 876–886.e874.
- 陈松、周洋、陈艳、顾坚(2018)。fastp:超快速全能型FASTQ预处理工具。生物信息学34,i884–i890。10.1093/bioinformatics/bty560。
- Dobin, A., Davis, C.A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M., Gingeras, T.R.(2013)。STAR:超快速通用RNA-seq比对工具。生物信息学29,15–21。10.1093/bioinformatics/bts635。
- McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., Garimella, K., Altshuler, D., Gabriel, S., Daly, M., DePristo,M.A.(2010)。基因组分析工具包:用于分析新一代DNA测序数据的MapReduce框架。基因组研究20,1297–1303。10.1101/gr.107524.110。
- Brouard, J.S., Schenkel, F., Marete, A., Bissonnette, N.(2019)。GATK联合基因分型工作流适用于RNA-seq实验中的变异检测。动物科学与生物技术杂志10,44。10.1186/s40104-019-0359-0。
- 廖玉、Smyth, G.K.、石文(2014)。featureCounts:用于将测序读数分配给基因组特征的高效通用程序。生物信息学30,923–930。10.1093/bioinformatics/btt656。
- Love, M.I., Huber, W., Anders, S.(2014)。基于DESeq2的RNA-seq数据折叠变化与离散度的稳健估计。基因组生物学15,550。10.1186/s13059-014-0550-8。
- Ihaka, R., Gentleman, R.(1996)。R:一种用于数据分析和图形的语言。计算与图形统计学杂志5,299–314。
- Risso, D., Ngai, J., Speed, T.P., Dudoit, S.(2014)。使用对照基因或样本的因子分析对RNA-seq数据进行标准化。自然生物技术32,896–902。10.1038/nbt.2931。
- Robinson, M.D., McCarthy, D.J., Smyth, G.K.(2010)。edgeR:用于数字基因表达数据差异表达分析的Bioconductor软件包。生物信息学26,139–140。10.1093/bioinformatics/btp616。
- 余光、王路广、韩珉、何庆瑜(2012)。clusterProfiler:用于比较基因簇间生物学主题的R软件包。组学16,284–287。10.1089/omi.2011.0118。
- Krämer, A., Green, J., Pollard, J., Jr., Tugendreich, S.(2014)。Ingenuity通路分析中的因果分析方法。生物信息学30,523–530。10.1093/bioinformatics/btt703。
- 顾祖光(2022)。复杂热图可视化。Imeta 1, e43。10.1002/imt2.43。
- Blighe, K., Rana, S., Lewis, M.(2019)。EnhancedVolcano:具备增强着色与标注功能的出版级火山图绘制工具。R软件包版本1.2.0。R软件包版本1.16。
¶ 图例
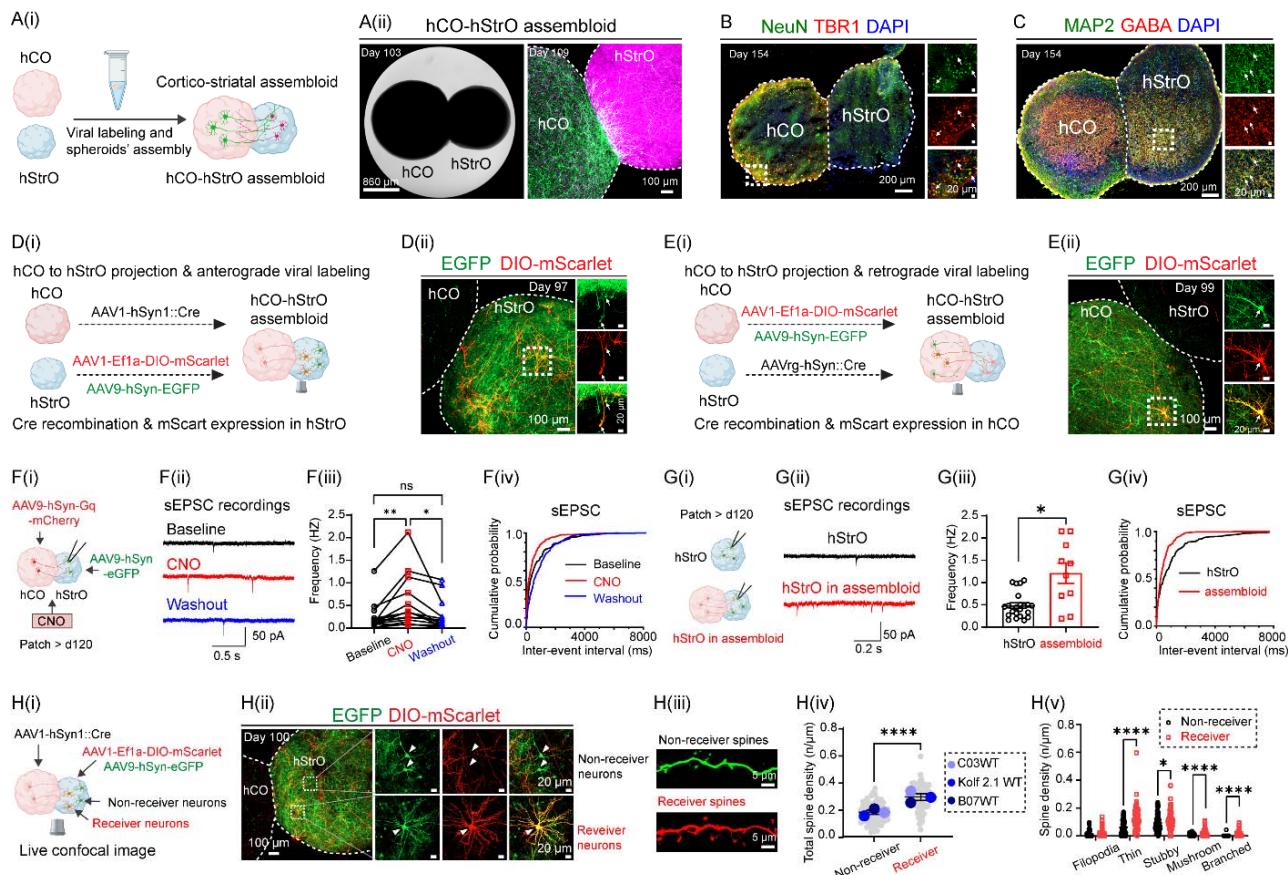
¶ 图 1. 皮质-纹状体类组装体的生成、连接图谱绘制与功能表征。另见图 S1。
(A)皮质-纹状体装配体的建模。(A(i))示意图展示了人皮质类器官(hCO)与纹状体类器官( )融合,在体外生成皮质-纹状体装配体的过程。(A(ii))融合hCO-hStrO装配体的明场图像(左,比例尺: )以及显示hCO(绿色)与hStrO(品红色)形态结构的共聚焦荧光图像(右,比例尺: )。(B, C) 装配体的神经元组成。
(B) 第154天NeuN(绿色)与TBR1(红色)免疫染色结果,虚线标记hCO-hStrO边界。右侧插图突出显示hCO中TBR1+/NeuN+ 的共定位情况。DAPI(蓝色)标记细胞核。比例尺:200微米(主图),20微米(插图)。 个组装体,来源于两个hiPSC细胞系,结果一致。
© 第154天hCO-hStrO组装体中MAP2(绿色)和GABA(红色)的免疫染色。右侧插图突出显示了hStrO中的GABA能神经元。 个组装体来自两个hiPSC细胞系,结果相似。(D, E) 利用病毒示踪技术进行连接映射。
(D) 顺行性追踪。(D(i)) 顺行性病毒追踪策略示意图。向hCOs注射AAV1-hSyn1-Cre病毒,向hStrOs注射AAV1-Ef1a-DIO-mScarlet和AAV9-hSyn-EGFP病毒。AAV1-hSyn1-Cre沿顺行方向通过跨突触机制从hCO末梢传递至hStrO神经元,诱导mScarlet表达,而EGFP作为纹状体分区标记物。(D(ii)) 显示接收皮质输入的纹状体神经元中EGFP+(绿色)与Cre依赖性mScarlet+(红色)的组装体共聚焦图像。n= 3个组装体获得类似结果(KOLF2.1J与A11 WT hiPSC细胞系)。
(E) 逆向追踪。(E(i))使用 AAVrg-hSyn-Cre 进行逆向标记示意图。(E(ii))共聚焦图像显示,标记有 AAV1-Ef1a-DIO-mScarlet 和 AAV9-hSyn-EGFP(绿色)的 hCO 神经元投射至 ,并接收逆向传输的 AAVrg-hSyn-Cre,从而诱导 mScarlet 表达(红色)。 个组装体具有类似结果(KOLF2.1J 和 A11 WT hiPSC 细胞系)。
(F) 化学遗传学激活可诱导突触传递。(F(i)) hCO 化学遗传学激活与 膜片钳记录的示意图。hCO 用 AAV9-hSyn-Gq-mCherry 转导,而 用 AAV9-hSyn-EGFP 标记。对 神经元进行全细胞膜片钳记录,并应用 CNO 激活 hCO 中的 Gq 信号通路。(F(ii)) hStrO 神经元在基线(黑色)、CNO(红色)和洗脱(蓝色)条件下的代表性自发性兴奋性突触后电流(sEPSC)轨迹。(F(iii))sEPSC 频率量化及 (Fiv) sEPSC 事件间期累积概率。( 个细胞,来自 6 个组装体,3 个 WT细胞系:C03, A11, KOLF2.1J)。采用盖瑟-格林豪斯校正的混合效应分析,图基多重比较检验。ns:无显著性, , 值 。**
(G) 对比单一hStrO与hCO-hStrO组装体的sEPSC记录。(G(i)) 分别从单一hStrO(黑色)或组装体中的hStrO(红色)记录sEPSC的示意图。(G(ii)) 代表性sEPSC波形及(G(iii)) sEPSC频率量化分析(数据来自3个单一hStrO的 个细胞、3个hCO-hStrO组装体的 个细胞)。非配对韦尔奇t检验: 0.05。(G(iv)) sEPSC事件间隔累积概率分布图。
(H) 树突棘分析的实时共聚焦成像。(H(i)) hStrO中接收神经元与非接收神经元的示意图。hCO用AAV1-hSyn1-Cre转导,而hStrO用AAV1-Ef1a-DIO-mScarlet和AAV9-hSyn-EGFP标记。(H(ii)) 显示非接收(仅EGFP+)和接收(EGFP+/mScarlet+,黄色)hStrO神经元的共聚焦图像。(H(iii)) 来自非接收(绿色,上图)和接收(红色,下图) 神经元的代表性树突棘。(H(iv)) 总树突棘密度定量,比较来自三个WT hiPSC细胞系(C03、KOLF2.1J、B07)的9个hCO-hStrO组装体中非接收神经元(灰色, 个树突)与接收神经元(红色, 个树突)。 (曼-惠特尼检验)。(H(v)) 树突棘亚型分类(丝状伪足型、细长型、粗短型、蘑菇型、分支型)。 ,****p (多重曼-惠特尼检验)。
数据以均值 标准误差表示。
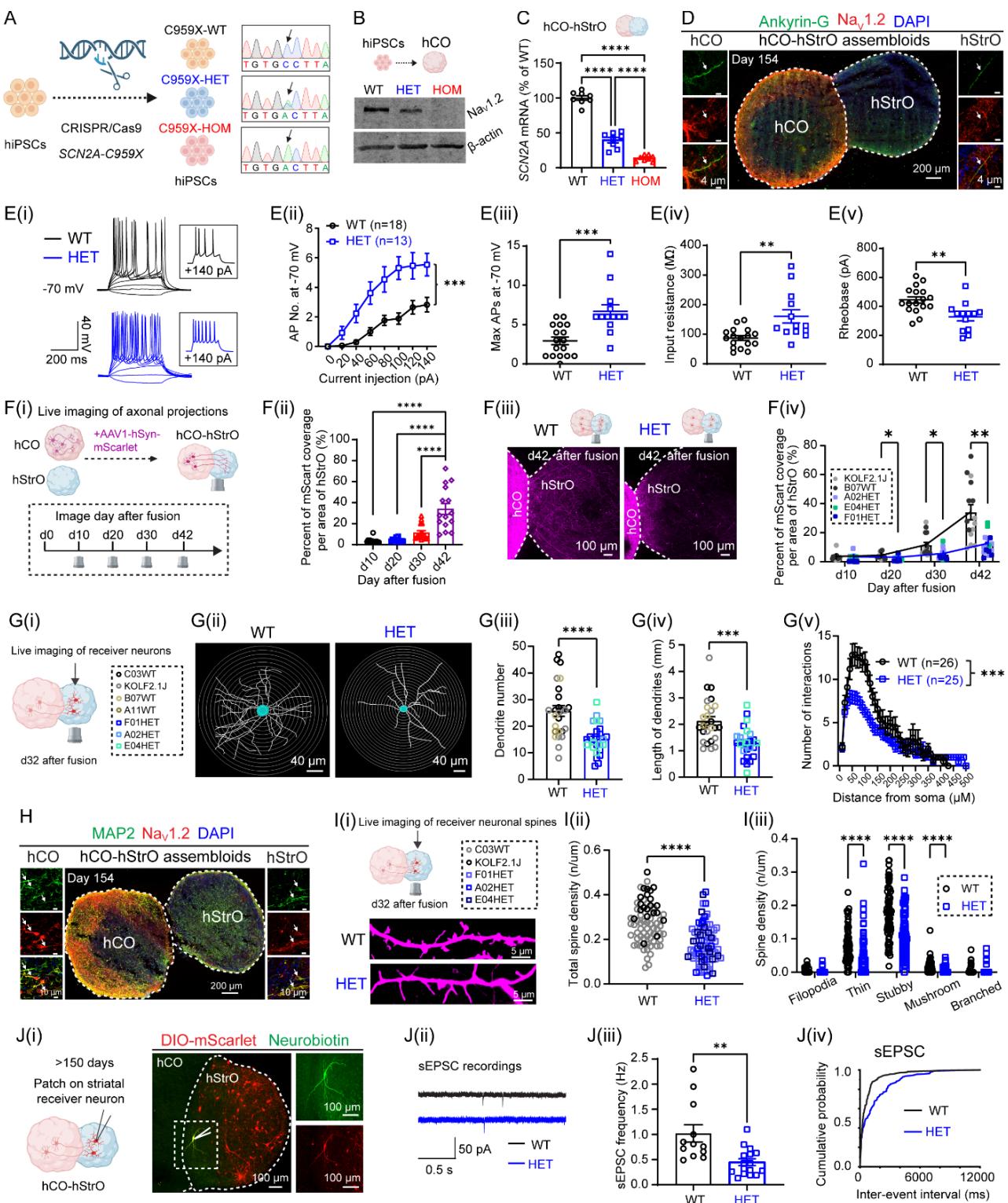
¶ 图 2. 携带 SCN2A-C959X 突变的人类组装体表现出受损的皮质-纹状体神经回路。另见图 S2。
(A) CRISPR/Cas9 编辑技术在人iPSCs中引入了SCN2A p.C959X突变。Sanger测序确认编辑成功:野生型(上图)保留胞嘧啶(C),杂合型(中图)显示重叠峰和同型变异(底部)显示完全性C-to-A替换,引入了提前终止密码子(X)。
(B)Western印迹显示WT、HET和HOM hCOs中NaV1.2(SCN2A)的表达情况,以 -actin作为内参。
© hCO-hStrO组装体中SCN2A mRNA水平显示,与WT相比,HET和HOM SCN2A-C959X突变体的表达降低。 个组装体/组,来自3个hiPSC细胞系(WT:KOLF2.1J、B07、C03;HET:A02、E04、F01;HOM:A03、D06、F03)。单因素方差分析结合Tukey检验,。
(D) 第154天hCO-hStrO组装体中锚蛋白G(绿色)与 (红色)的免疫染色,显示 定位于轴突起始段(AIS)。 个组装体。
(E) 携带SCN2A-C959X突变的神经元电生理改变。(E(i)) 野生型(WT)和杂合型(HET)神经元在 V下的动作电位代表性波形。插图:对 电流注射的动作电位响应。(E(ii)) 在去极化电流脉冲( )下诱发的动作电位。( ,双因素方差分析)。(E(iii)) 在 下的单个及平均最大动作电位数量。曼-惠特尼检验, 。(E(iv)) 下的输入电阻及(E(v))基强度测量。非配对t检验。 。
(F) SCN2A-C959X 组装体中的轴突投射缺陷。(F(i)) 实验时间线及(F(ii)) 融合后第10、20、30和42天组装体内从hCO到hStrO的轴突投射量化。单因素方差分析,Tukey检验, 。(F(iii))第42天WT与HET组装体中轴突投射的代表性图像(mScarlet )。(F(iv)) hStrO区域在第10、20、30和42天的mScarlet覆盖量化,比较WT( 个组装体,来源B07、KOLF2.1J)与HET( 个⁺组装体,来源F01、A02、E04)。采用Geisser-Greenhouse校正的混合效应模型,Sidak检验。 .05, 。
(G) SCN2A-C959X 组装体中的树突形态缺陷。 (G(i)) hStrO中接收神经元鉴定的示意图。 (G(ii)) WT和HET中神经元形态(Sholl分析)的代表性图像。 (G(iii)) 树突总数(非配对t检验),(G(iv)) 树突长度(非配对t检验),以及 (Gv) WT( 个神经元,B07、C03、A11、KOLF2.1J细胞系)和HET( 个神经元,F01、A02、E04细胞系)中的交互数(双向ANOVA)。 ,p值 。
(H) 树突中NaV1.2的定位。hCO-hStrO组装体中MAP2(绿色)、NaV1.2(红色)和DAPI(蓝色)的免疫染色结果,证实了NaV1.2在神经元树突中的表达。数据来源于两个hiPSC系中的n=3个组装体,结果一致。
(I) SCN2A-C959X 组装体中树突棘缺陷。(I(i)) 野生型和杂合型纹状体接收神经元(mScarlet )树突棘的共聚焦图像。(I(ii)) 野生型(灰色,n = 74个树突,9个组装体,C03、KOLF2.1J、B07细胞系)和杂合型(蓝色)接收神经元总树突棘密度的量化。
n = 86 个树突,9 个组装体,F01、A02、E04 细胞系)。Mann-Whitney 检验,****p < 0.0001。(I(iii))脊柱亚型分析。 (多重 Mann-Whitney 检验)。
(J) SCN2A-C959X组装体中突触传递缺陷。(J(i)) hCO-hStrO组装体hStrO中接收神经元(mScarlet ,红色)的共聚焦图像。神经生物素标记的膜片钳神经元(绿色)证实被记录神经元为接收神经元。(J(ii)) WT(黑色,上图)与HET(蓝色,下图)hStrO接收神经元sEPSC示例波形。(J(iii)) WT 个细胞,7个组装体,A11、KOLF2.1J细胞系)与HET 个细胞,7个组装体,A02、E04细胞系)sEPSC频率。非配对t检验, 。(J(iv)) sEPSC事件间隔累积概率分布。
数据以平均值 标准误表示。
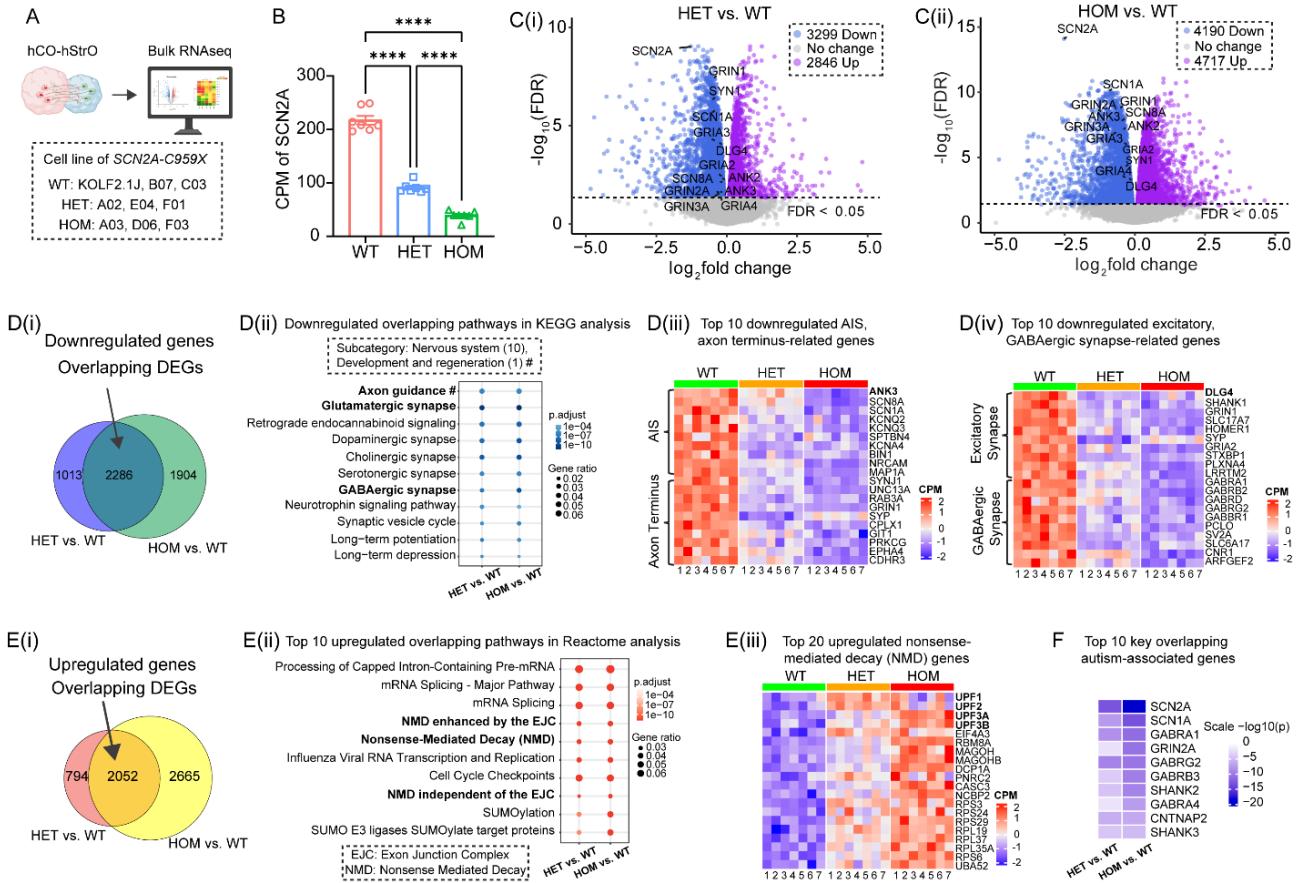
¶ 图 3. 转录组学分析揭示携带 SCN2A-C959X 突变的类组装体中存在差异基因表达和通路改变。另见图 S3。
(A) 实验流程。每组 个组装体来自WT(iPSC细胞系:KOLF2.1J、B07、C03), 杂合(iPSC系:A02、E04、F01)与纯合(iPSC系:A03、D06、F03)。
(B)条形图显示批量RNA测序中WT、HET和HOM组SCN2A表达的每百万计数(CPM)。采用单因素方差分析与Tukey事后比较, 。
© 差异基因表达分析。火山图展示HET与WT(C(i))及HOM与WT(C(ii))比较中的差异表达基因(DEGs)。上调基因(洋红色)、下调基因(蓝色)及无显著差异基因(灰色)(错误发现率 )。图中标注了与轴突及突触通路相关的关键下调基因。
(D) 下调基因通路。 (D(i)) 维恩图展示 HET 与 WT 及 HOM 与 WT 比较中重叠的下调基因。 (D(ii))神经系统及发育与再生类别(标有“#”)中下调基因的 KEGG 通路富集分析。 (D(iii)) 轴突缺陷。排名前列的下调轴突相关基因热图,分为轴突起始节段(AIS)(前10位)与轴突末端基因(前10位),显示 HET 与 HOM 中的轴突缺陷。 (D(iv)) 突触缺陷。排名前列的下调突触相关基因热图,分为兴奋性(前10位)与抑制性(GABA能,前10位)突触。
(E) 上调基因通路。(E(i)) 韦恩图显示HET vs. WT与HOM vs. WT中重叠的上调基因。(E(ii)) 对前10个重叠上调通路进行Reactome通路富集分析,关键通路与无义介导的mRNA降解相关。衰减(NMD)以粗体标出。(E(iii))HET与HOM相对于WT组中前20个上调NMD相关基因的热图(CPM:每百万计数)。
(F)自闭症相关基因改变。HET相对于WT及HOM相对于WT组中前10个ASD相关差异表达基因(DEGs)的热图。标度: 。
¶ 补充信息
¶ 补充图 1
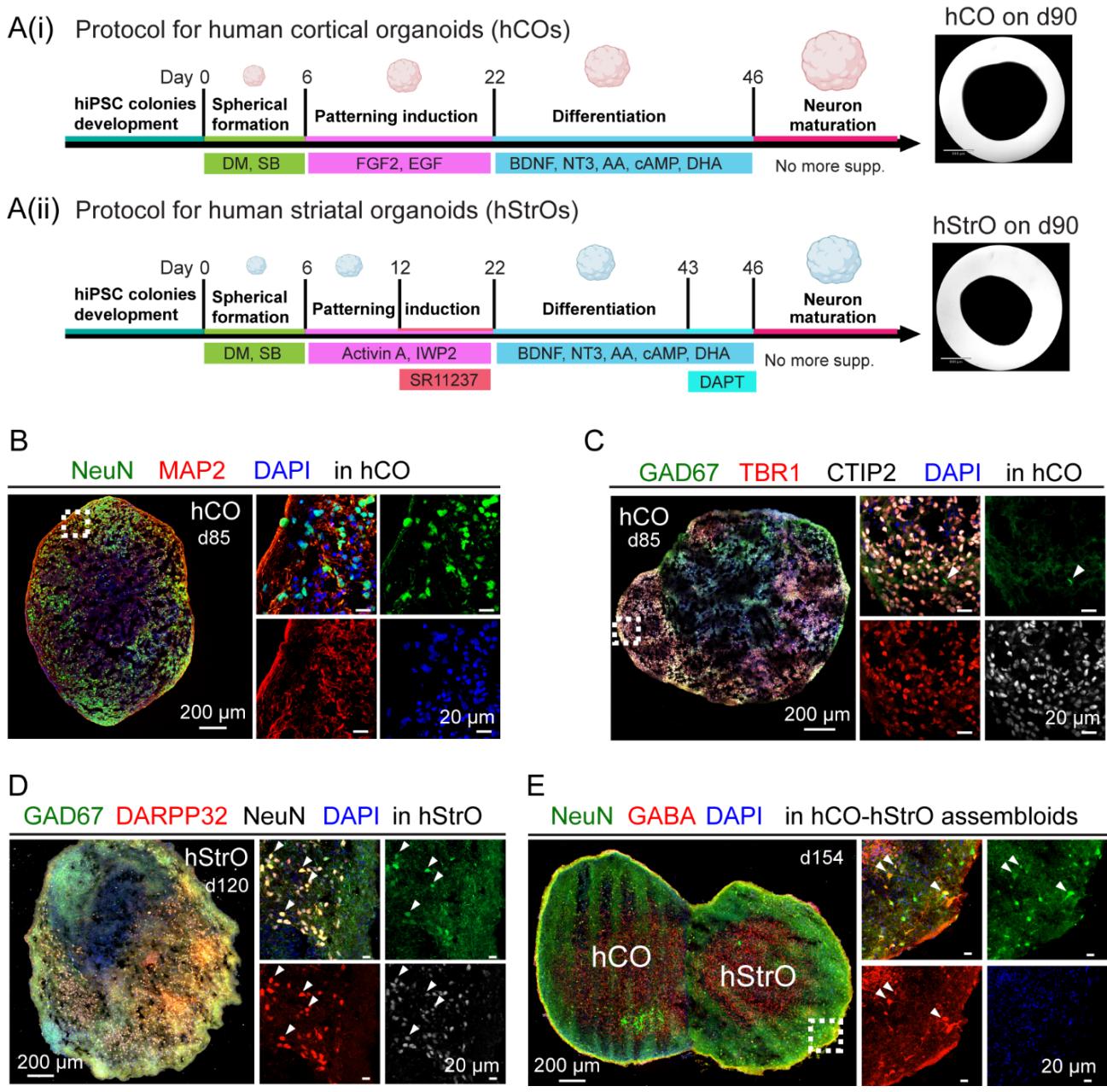
¶ 图 S1. 人脑皮质和纹状体类器官的生成与表征。与图1相关
(A(i)) 人皮层类脑器官(hCOs)的培养方案。图示为人皮层类脑器官的分化时间线:人诱导多能干细胞(hiPSCs)依次经历类脑器官形成(第6天)、模式诱导(第22天)、分化(第46天)及神经元成熟阶段。关键添加成分包括双重SMAD抑制剂(DM/SB)、成纤维细胞生长因子2(FGF2)、表皮生长因子(EGF)、脑源性神经营养因子(BDNF)、神经营养因子3(NT3)、抗坏血酸(AA)、环磷酸腺苷(cAMP)和二十二碳六烯酸(DHA)。
右图:hCO 的明场图像,比例尺:860 微米。(Aii)人类纹状体类器官(hStrOs)制备流程。采用相似的分化时间线,并添加模式诱导因子(包括 Activin A、Wnt 信号通路抑制剂 IWP2 及 SR11237),随后进行分泌酶抑制剂DAPT用于晚期分化。右图:hStrO的明场图像,比例尺: 。
(B) 第85天人脑类器官神经元标记物免疫染色结果,显示神经元核蛋白(NeuN,绿色,成熟神经元标记物)、微管相关蛋白2(MAP2,红色,树突标记物)以及4′,6-二脒基-2-苯基吲哚(DAPI,蓝色,细胞核标记物)。高倍放大图像展示各标记物在球状体内的具体定位。
© hCO中的神经元类型表征。免疫染色显示谷氨酸脱羧酶67(GAD67,绿色,GABA能神经元标记物)呈稀疏表达,而T-box脑转录因子1(TBR1,红色,深层皮质神经元标记物)、COUP-TF相互作用蛋白2(CTIP2,白色,皮层下投射神经元标记物)及DAPI(蓝色)均呈强表达。高倍放大图像展示了球状体内不同的神经元群体。
(D) 第120天hStrO的免疫染色显示GAD67(绿色)、多巴胺和cAMP调节磷蛋白32(DARPP32,红色,纹状体投射神经元标记物)、NeuN(白色)和DAPI(蓝色)。高倍放大图像凸显了这些神经元标记物的空间分布。
(E) hCO-hStrO组装体的免疫染色结果,显示NeuN(绿色)、γ-氨基丁酸(GABA,红色,作为GABA能神经元的标志物)和DAPI(蓝色)。皮质区和纹状体区分别标记为hCO和hStrO。高放大倍数图像展示了融合位点的标志物定位,表明皮质与纹状体神经元群体已实现整合。
¶ 补充图 2
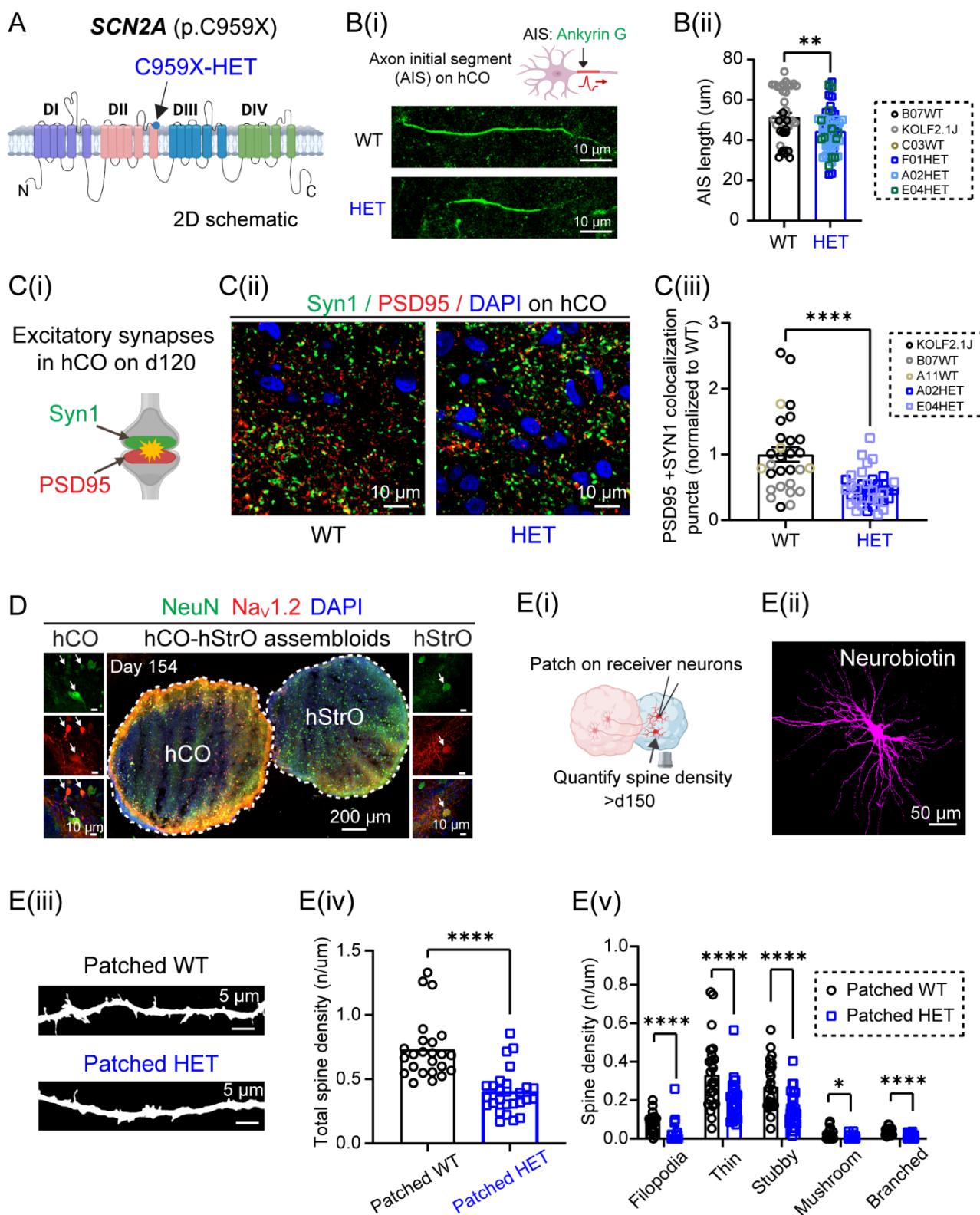
¶ 图 S2。SCN2A 缺陷破坏 hCO-hStrO 组装体中 AIS 长度、突触连接及树突棘形态。与图 2 相关
(A) SCN2A 示意图,标示出 p.C959X 无义突变位点。
(B) 轴突起始段形态。(B(i)) WT与HET SCN2A-C959X神经元中由锚蛋白G(绿色)标记的轴突起始段示意图及代表性图像。(B(ii)) 轴突起始段长度量化(WT:n = 42个神经元,来自B07、C03、KOLF2.1J hiPSC细胞系;HET:n = 56个神经元,来自F01、A02、E04 hiPSC细胞系)。非配对韦尔奇t检验, $* * _ { \mathrm { p } } < 0 . 0 1 $ 。
© SCN2A突变hCOs中兴奋性突触形成受损。(C(i)) 由Syn1(绿色,突触前)和PSD95(红色,突触后)标记的兴奋性突触示意图。(C(ii)) WT和HET SCN2A-C959X hCOs中兴奋性突触(PSD95+/Syn1+共定位)的代表性图像。比例尺: 。(C(iii)) 兴奋性突触定量分析。WT:来自9个类器官的29张图像(KOLF2.1J, B07, A11);HET:来自10个类器官的37张图像(A02, E04)。非配对t检验, 。
(D)hCO-hStrO组装体中NeuN(绿)、 (红)和DAPI(蓝)的免疫染色,显示 与NeuN在hCO和hStrO中均存在共定位,表明 在神经元胞体中的表达。
(E) hCO-hStrO组装体切片中SCN2A突变型纹状体神经元的树突棘密度降低。(E(i)) hStrO接收神经元上进行膜片钳记录及树突棘密度定量分析的示意图。(E(ii)) 神经生物素标记(品红色)的膜片钳记录神经元的代表性图像。(E(iii)) 膜片钳记录神经元树突棘的代表性图像。(E(iv)) 接收神经元总树突棘密度的定量分析。WT(灰色, 个树突,来自4个组装体,A11,KOLF2.1J);SCN2A-C959X杂合突变型(蓝色, 个树突,来自5个组装体,A02、E04)。曼-惠特尼检验:****p < 0.0001。(E)树突棘亚型分析(丝状伪足型、细长型、短粗型、蘑菇型、分枝型)。多重曼-惠特尼检验, ,****p<0.0001。
数据以平均值±标准误表示。
¶ 补充图 3
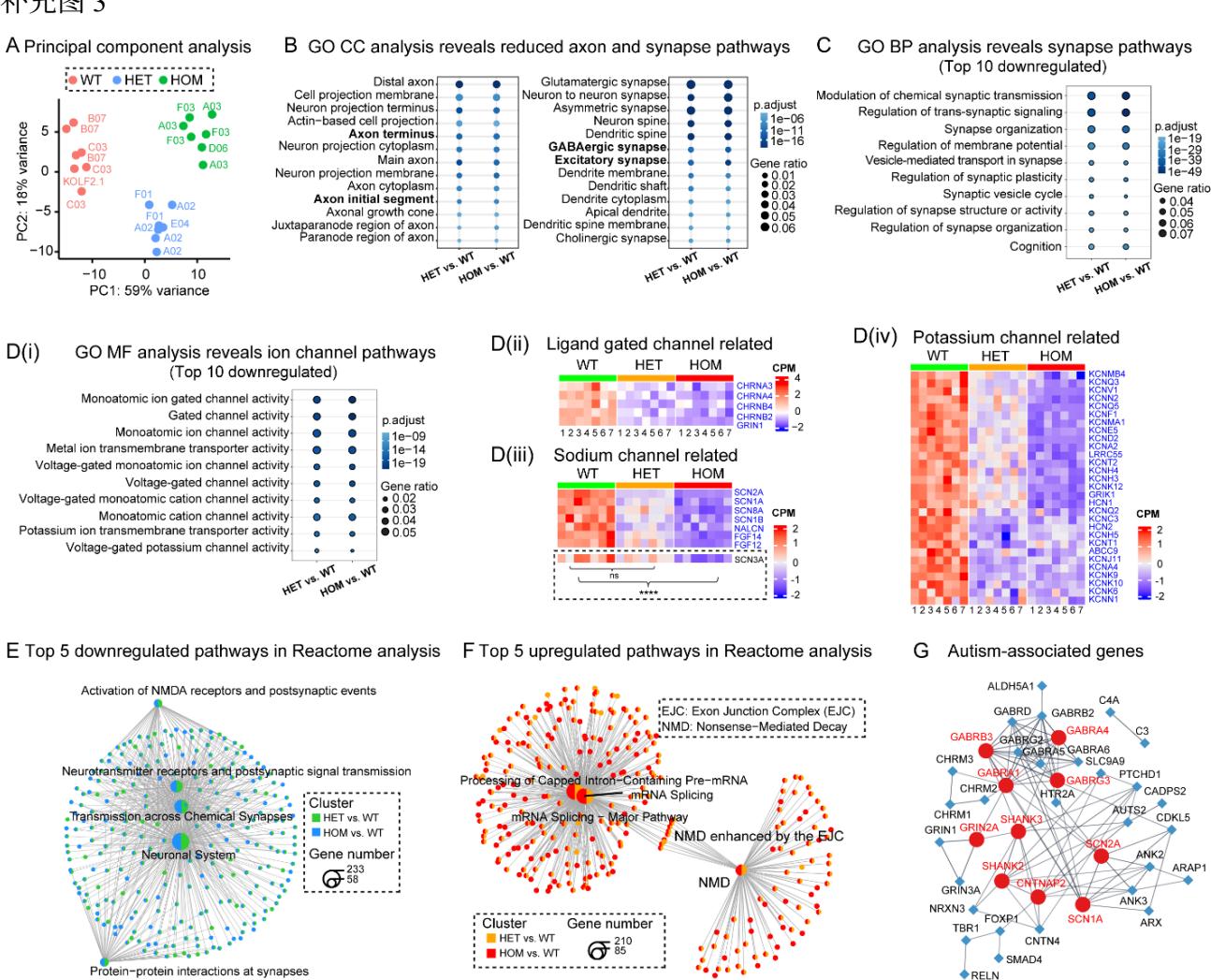
¶ 图 S3. SCN2A缺陷型组装体中的基因表达失调和通路改变。与图 3 相关。
(A) 全局转录组改变。基于整体基因表达水平的转录组数据主成分分析显示,WT(KOLF2.1J、B07、C03系)、HET(A02、E04、F01系)和HOM(A03、D06、F03系)类器官集合呈现聚类分布。每个点代表一个样本(每组n 个类器官集合)。
(B) 重叠下调基因的基因本体(GO)细胞组分(CC)分析,显示轴突末梢和轴突起始段;兴奋性突触以及GABA能突触的减少。
© SCN2A-C959X组装体中受破坏的突触相关通路。前10个重叠的HET与WT及HOM与WT比较中GO生物学过程的下调通路比较。
(D) SCN2A-C959X 类组装体中离子通道相关通路紊乱。(D(i)) GO分子功能(MF)中前10个重叠下调通路揭示了杂合与纯合类组装体中的离子通道基因失调。重叠下调离子通道基因的热图(蓝色显示),分为配体门控通道相关(D(ii))、电压门控钠通道相关(D(iii))及电压门控钾通道相关(D(iv))。突出显示的SCN3A在HET组中未显示显著变化,但在HOM组中显著降低。
(E) HET与HOM组装体中的突触功能障碍。下调差异表达基因中富集的前5条Reactome通路网络图,主要影响NMDA受体活性、神经递质信号传导、突触传递及神经元相互作用,表明SCN2A缺陷组装体中存在突触功能紊乱。
(F) SCN2A-C959X类器官中RNA监视增强。上调差异表达基因中最富集的5条Reactome通路网络图揭示了其与mRNA剪接、无义介导的mRNA降解(NMD)以及外显子连接复合体(EJC)功能的关联。
(G) ASD相关基因的网络分析,描绘了SCN2A突变类组装体中差异表达的ASD相关基因之间的相互作用。排名前十的枢纽基因以红色高亮显示。
¶ 人诱导多能干细胞系
通过CRISPR/Cas9介导的基因组编辑34,35技术,在早期传代(p2)的KOLF2.1J参考iPSCs36中构建了SCN2A c.2877C>A(p.Cys959Ter)突变型hiPSC细胞系。我们重新进行了多能性检测和基因组完整性评估,以确保hiPSC37的质量。本研究使用了四种同源野生型对照细胞系(KOLF2.1J、C03、B07和A11)、三种SCN2A杂合突变细胞系(A02、E04和F01)以及三种SCN2A纯合突变细胞系(A03、D06和F03)。每一批次hiPSC来源的类器官或组装体均通过Sanger测序和基因分型确认了突变的成功引入。
¶ 抗体
对于免疫染色,使用的一抗包括Anti-NeuN(鸡源,GeneTex,GTX00837,1:1000;兔源,Cell Signaling,24307S,1:1000)、Anti-TBR1(兔源,Abcam,ab31940,1:300)、Anti-MAP2(鸡源,Novus Biologicals,NB300-213,1:1000;鼠源,Millipore,MAB378(1:200)、抗GABA抗体(兔源,Sigma-Aldrich,A2052,1:1000)、抗GAD67抗体(小鼠源,Sigma-Aldrich,MAB5406-25UG,1:100)、抗Ctip2抗体(大鼠源,Abcam,ab18465,1:300)、抗DARPP32抗体(兔源,Abcam,ab40801,1:200)、抗锚蛋白G抗体(小鼠源,AntibodiesInc.,75-146-020,1:200)、抗SCN2A抗体(兔源,Sigma-Aldrich,ZRB1300,1:200)、HA标签抗体(兔源,Cell Signaling,3724T,1:200)、突触素1抗体(小鼠源,Synaptic Systems,106011,1:800)、PSD-95抗体(兔源,Thermo Fisher,51-6900,1:800)。二抗包括山羊抗鸡Alexa Fluor647(Thermo Fisher,A32933,1:500)、山羊抗兔Alexa Fluor 647(A21244,1:500)、山羊抗小鼠Alexa Fluor 647(A21235,1:500)、山羊抗大鼠Alexa Fluor 555(A21434,1:500)、山羊抗兔Alexa Fluor 488(A11034,1:500)、山羊抗兔Alexa Fluor Plus 555(A32732,1:500)、山羊抗小鼠Alexa Fluor Plus 555(A32727,1:500)以及山羊抗小鼠Alexa Fluor 488(A11001,1:500)。
对于Western印迹分析,使用的一抗包括兔抗SCN2A(NaV1.2)(Alomone Labs,货号ASC-002,稀释比例1:200)、兔抗SCN2A(西格玛奥德里奇,货号ZRB1300,稀释比例1:200)以及鼠抗 -肌动蛋白(赛默飞世尔,货号MA5-15739,稀释比例1:1000)。使用的二抗为IRDye® 800CWβ山羊抗兔IgG(LI-COR,货号926-32211,稀释比例1:2500)和IRDye® 800CW山羊抗鼠IgG(货号926-32210,稀释比例1:5000)。
¶ 方法详情基因分型
收集hiPSCs、类器官及组装体并进行基因分型。使用组织DNA提取试剂盒(Macherey-Nagel,美国伯利恒)提取DNA。采用以下引物进行基因特异性PCR:正向(5’–3’):TTGAGACAGTTACCTGTACATTTGC;反向(5’–3’):TAATAGACAATAGGAAGTGGCCTTG。PCR反应体系( )包含PCR预混液、引物、模板DNA及无核酸酶水,热循环条件为: 预变性2分钟; 30秒、 30 秒、 1分钟,共35个循环; 终延伸10分钟后 保存。对700 bp PCR产物进行桑格测序,确认基因组编辑结果:野生型在突变位点保留胞嘧啶(C),杂合子系出现重叠峰,纯合子系实现完全C-to-A替换,从而引入提前终止密码子。
¶ 从hiPSCs生成hStrOs和hCOs。
¶ 三维神经类器官的生成
无饲养层hiPSCs集落被接种于Matrigel基质胶(康宁,#354230)上的StemFlex培养基(赛默飞,#A3349401)中,每日更换培养基。细胞每4-5天使用Versene解离液(赛默飞,#15040066)进行传代。质量控制评估包括桑格测序、核型分析和免疫细胞化学检测。未分化hiPSCs集落呈现正常均一的形态学特征,边缘轮廓清晰且自发分化程度极低。
类器官是依照改进的Muria方案15生成的。将hiPSCs使用Accutase(赛默飞,#NC9839010)消化为单细胞,以每孔10,000个细胞的密度接种于超低吸附96孔板(康宁,#CLS7007)中,培养基采用添加了 ROCK抑制剂Y27632(Selleck Chemicals,#S1049)的Essential 8培养基(赛默飞,#A1517001)。将平板以 离心3分钟,置于 、 条件下培养。24小时后,将类器官转移至Essential 6培养基(赛默飞,#A1516401),该培养基补充有 多索吗啉(Sigma-Aldrich,#P5499)、 SB-431542(R&D Systems,#1614)和 XAV-939(Tocris,#3748),连续培养5天并每日更换培养基。
¶ 模式形成与分化为hCO和hStrOs
第6天,将类器官转移至超低吸附6孔板(康宁,#3471),并加入含有Neurobasal-A(赛默飞,#10888022)、不含维生素A的B-27(赛默飞,#12587010)、GlutaMAX(1:100,赛默飞,#35050061)以及青霉素-链霉素(1:100,赛默飞,#15140122)的神经培养基。对于hCOs,该培养基额外添加20 ng/mL FGF2(R&D Systems,#233-FB-500)和 EGF(R&D Systems,#236-EG)。对于hStrOs,则补充2.5 M WNT抑制剂IWP-2(Selleck Chemicals,#S7085)和50 ng/mL重组Activin A(PeproTech,#120-14P)。第12天,向hStrOs中添加 SR11237(Tocris,#3411)。
¶ 神经元分化和成熟
从第22天起,通过向神经培养基中添加BDNF( ,PeproTech,#450-02)、NT-3( ,PeproTech,#450-03)、抗坏血酸( ,Wako,#323-44822)、二丁酰环腺苷酸( ,Santa Cruz,#sc-201567A)和DHA( ,MilliporeSigma,#D2534),将神经祖细胞分化为神经元(适用于hCO和hStrO)。在第42-45天期间,向hStrO培养基中添加2.5 M DAPT(Stemcell Technologies,#72082),同时保留BDNF、NT-3、抗坏血酸、cAMP和DHA。从第46天起,将细胞培养物维持于含有B-27 Plus补充剂(Thermo Fisher,#A3582801)的神经培养基中,每4-5天更换一次培养基。
¶ 皮层-纹状体类组装体的病毒标记与活细胞成像皮质-纹状体装配体的生成13
为生成皮层-纹状体装配体,在病毒标记后将一个hCO和一个hStrO转移至含1 mL NM培养基的 EP管中,于 孵育3-4天,并于第2天进行完全培养基更换。装配体形成后,使用剪口处理的P1000移液器将其转移至超低吸附板中以适应其尺寸。装配过程在分化第60-76天期间进行。
¶ 顺行性及逆行性示踪
在顺行标记实验中,第60天时,用AAV1-hSyn-Cre(Addgene #105553)标记hCOs,同时使用AAV1-Ef1a-DIO-mScarlet(Addgene #131002)和AAV9-hSyn-EGFP(Addgene )标记hStrOs。逆行标记实验中,则用AAV1-hSyn-Cre标记hStrOs,而使用AAV1-Ef1a-DIO-mScarlet和AAV9-hSyn-EGFP标记hCOs。组装体在培养过程中每4天更换培养基。1个月后,将标记后的组装体转移至玻璃底24孔板(Cellvis, #P24-0-N)中,置于BrainPhys培养基(STEMCELL, #5790)内,于 、 CO2条件下平衡15分钟后,使用蔡司LSM 900共聚焦显微镜( 物镜,Z轴层扫)进行成像。
¶ 轴突投射成像
为评估从hCO到hStrO的投射情况,于第60天用AAV1-hSyn-mScarlet(Addgene #131001)标记hCO,在第65天与hStrO组装,并在融合后第10、20、30和42天使用蔡司LSM 900( 物镜,0- 深度,Z轴堆叠扫描)进行成像。轴突投射通过使用Fiji软件(https://fiji.sc/)测量hStrO中mScarlet覆盖百分比进行量化。
¶ 接收神经元与非接收神经元标记
hCOs使用AAV1-hSyn-Cre进行标记,而hStrOs在第60天使用AAV1-Ef1a-DIO-mScarlet和AAV9-hSyn-EGFP标记,随后于第65日进行融合。一个月后,在 、 条件下平衡15分钟,使用蔡司LSM 900( 物镜, 倍变焦,Z轴层扫)进行树突棘成像。EGFP+/mScarlet– 神经元被归类为非接收神经元,而mScarlet+神经元则被指定为接收神经元。每个组装体至少选取三处树突,通过Neurolucida 360和Neurolucida Explorer(MBF Bioscience)进行树突棘密度与形态分析。
¶ 接收神经元中的树突棘成像
人类皮层类器官在第60天用AAV1-hSyn-Cre标记,而人类纹状体类器官在第60天用AAV1-Ef1a-DIO-mScarlet标记,随后在第65天进行融合。一个月后,组装类器官被转移到BrainPhys培养基中,并在 微米深度对受体(mScarlet+)神经元进行成像。使用 倍物镜和 倍变焦观察树突棘,并通过Neurolucida 360和Neurolucida Explorer(MBF Bioscience)进行Sholl分析,评估神经元复杂度、树突棘密度及形态。
¶ 膜片钳记录
¶ 急性脑片制备
从3-6月龄的脑类器官和hCO-hStrO组装体中制备切片。类器官和组装体被快速包埋于 琼脂糖的切片溶液中,该溶液成分包含(单位mM):110 胆碱氯化物、 、 、25 、 、 、25 葡萄糖、1 抗坏血酸钠以及3.1 丙酮酸钠( , ,通入 和 饱和)。使用振动切片机(德国Leica VT1200 S)制备冠状切片(厚度20 )。切片在 的切片溶液中孵育10分钟,随后转移至人工脑脊液(aCSF;成分mM: Cl、 、 、 、 、 、10 葡萄糖; ,305–315 mOsm,通入 和 饱和),在 下维持10–20分钟后,于室温保存至少30分钟再进行记录。
¶ 装配体切片中的全细胞电生理学
脑片置于持续灌流aCSF ,流速 的记录槽中。神经元通过配备IR-2000相机(Dage-MTI)的红外微分干涉相差显微镜(Olympus BX-51WI)进行观察。在纹状体中等棘神经元(MSNs)和皮质锥体神经元上实施胞体全细胞膜片钳记录。MSNs通过其特征性形态(包括中等尺寸、多角形/树突棘状胞体)进行识别,而皮质锥体神经元则以显著的主树突为区分标志。使用P-1000拉制仪(Sutter Instruments)制备开放尖端电阻为 的薄壁硼硅酸盐玻璃微管(BF150-110-10,Sutter Instruments)。
采用电压钳记录自发性兴奋性突触后电流(sEPSCs)以评估突触活动。细胞内液成分为(单位mM):120 甲磺酸铯3、 、0.2 EGTA、10 HEPES、 、0.3 Tris3-GTP、14 Tris2-磷酸肌酸,并用CsOH调节至 (渗透压295–305 mOsm)。在膜电位钳制于 的条件下,以无缝记录模式采集3分钟sEPSCs信号。
记录使用Axon MultiClamp 700B放大器(Molecular Devices)进行,数据通过pClamp 11.4软件采集,经 滤波后,以 采样率通过Axon Digidata 1550B加HumSilencer数字化仪(Molecular Devices)采样。串联电阻(Rs)维持在 ,不稳定Rs记录( )被排除。数据文件以ABF 1.8格式保存,并使用Mini Analysis程序(v6.08版,Synaptosoft)进行分析。
在化学遗传学实验中,hCOs在60天时用AAV9-hSyn-hM3D(Gq)-mCherry(Addgene #50474)标记,而hStrOs在60天时用AAV9-hSyn-EGFP标记,随后于第65天进行组装。两个月后,对组装体进行sEPSC记录。选择hStrO中被mCherry标记轴突包围的EGFP MSNs进行记录。先记录3分钟基线活动,接着进行 CNO的浴式给药3分钟,最后进行3分钟的冲洗记录。
细胞标记的内部溶液含有 的神经生物素。记录结束后( 分钟),将切片在 多聚甲醛( )中于室温固定20–30分钟,用PBS清洗,并在 下与Alexa 647偶联链霉亲和素(封闭液按1:250稀释)38共孵育过夜。对已记录的神经元进行神经元形态(包括树突棘)成像,采用蔡司L 显微镜完成。
¶ 离体神经元全细胞电生理学
采用改良型Worthington木瓜蛋白酶解离试剂盒(木瓜蛋白酶解离系统,Worthington,LK003150)将脑类器官解离为单细胞。随机选取 个成熟人脑类器官( 日龄)转移至60毫米培养皿,吸除多余培养基后加入5毫升木瓜蛋白酶-脱氧核糖核酸酶混合溶液。将组织剪碎成 毫米大小的碎片,于 、 条件下以80转/分钟振荡孵育30分钟,轻柔吹打后再继续孵育10分钟。将所得悬浮液转移至15毫升锥形管,待碎片沉降后取上清液与抑制剂溶液混合, 离心7分钟。沉淀重悬于神经基础培养基,经40 米细胞筛(CELLTREAT Scientific Products, #229481)过滤后计数。取约 个细胞接种于24孔板中12毫米圆形盖玻片(Neuvitro, #GG-12-1.5-Pre)上。神经元在含B-27添加剂(不含维生素A)的神经基础培养基中培养7天,隔天更换半数培养基。从第7天起,使用含B-27 Plus添加剂的神经基础培养基维持培养,每周进行两次半数培养基更换。
在全细胞电流钳记录中,内部溶液包含(单位 mM):122 甲基硫酸钾 、4 氯化钾、2 氯化镁 、0.2 EGTA、10 HEPES、4 三磷酸腺苷钠2、0.3 三磷酸鸟苷三羟甲基氨基甲烷盐和 14 三羟甲基氨基甲烷磷酸肌酸,用 KOH 调节至 ,渗透压 。记录使用 Axon MultiClamp 700B 放大器(Molecular Devices 公司)进行,采样频率为 ,并通过 pClamp 11.4 软件以 滤波。通过一系列 400 毫秒的超极化和去极化电流阶跃(从 至 ,以 递增),在正常静息膜电位(RMP)或 条件下进行 5 秒扫描,评估了下垂比、输入电阻和放电特性。
输入电阻 的计算公式为:
输入电阻 (电压 baseline−电压 steady-state) (兆欧)其中Vbaseline为静息膜电位或 ,Vsteady-state是在 刺激结束前记录的电压。
高密度多电极阵列(HD-MEA)对皮层-纹状体组装体的记录高密度微电极阵列制备与类器官组装接种HD-MEA(3Brain,6孔板)用 Tergazyme(每孔 , ,1小时)清洗,超纯水漂洗三次, 乙醇消毒1小时,干燥后于室温PBS中孵育过夜。随后依次用多聚左旋鸟氨酸( ;Sigma-Aldrich,#P4957)包被过夜并清洗,再用层粘连蛋白( 50 μg/mL,37 ℃ 条件下孵育 ≥ 2小时 )进行二次包被。
皮质-纹状体(hCO-hStrO)组装体(~月龄)使用宽口移液管转移至HD-MEA芯片上,静置2小时使其沉降。为促进贴壁,每2小时添加20mL培养基,持续8小时,次日更换 培养基。培养体系采用添加N2、B27 Plus、50 M cAMP及 抗坏血酸的BrainPhys培养基,每两周换液一次。培养2周后进行电生理记录。
¶ 电生理记录与分析
记录当天更换培养基后,将培养板置于 、 条件下孵育15分钟。随后使用HyperCAM Alpha多孔板系统配合Brainwave V软件(v5.6,3Brain Switzerland)进行5分钟细胞外电活动记录。记录采用 电极的高密度微电极阵列(电极间距 ,记录区域面积 ,采样率为 ,并施加 高通滤波及 带通滤波。频谱分析采用汉明窗的快速傅里叶变换进行处理。
使用8.0标准偏差阈值检测尖峰信号,峰值寿命为2.0毫秒,不应期为1.0毫秒,峰前波持续时间为1.0毫秒。尖峰频率低于 赫兹(5次/分钟)的电极被排除。将连续 个尖峰且峰间间隔小于 100毫秒的信号识别为爆发,而网络爆发通过基于募集率的算法检测,要求电极激活率超过 且最小爆发规模为50个尖峰。使用主成分分析(PCA,3个成分)和基于间隙统计量的K-Means聚类进行尖峰排序。
¶ 低温保护、免疫细胞化学与成像分析
¶ 样品制备
脑器官体和组装体在 下用 多聚甲醛(PFA)的PBS溶液固定过夜,用PBS清洗后转入 蔗糖-PBS溶液浸泡2-3天直至完全沉底。随后将样本在最佳切割温度包埋剂
(OCT)化合物 (Tissue-Tek, 4583, Sakura Finetek) 和 蔗糖-PBS处理后再进行包埋。经过包埋后,使用Leica CM1850冷冻切片机获得冷冻切片(厚度20–40 μm)。置于玻璃盖玻片上的二维培养切片在室温下用 多聚甲醛固定20分钟。
¶ 免疫染色39
冷冻切片用PBS清洗( ,5分钟),进行透化处理,并在室温下用含 Triton X-100和5 正常山羊血清的PBS,或含 Tween-20的 Block-Ace(大日本住友制药,UK-B80)封闭1小时。一抗于 孵育过夜后,用PBS清洗( ,10分钟)。样品随后在封闭缓冲液中与Alexa Fluor标记的二抗于室温孵育1小时。切片用含DAPI的抗淬灭封片剂(VECTASHIELD, H-2000)封片,并用玻璃盖玻片密封。图像使用配备空气扫描模块的LSM900共聚焦荧光显微镜(卡尔蔡司,耶拿,德国)采集。
¶ 轴突起始段(AIS)长度量化23
图像采集使用 油浸物镜,每个类器官至少选取三个区域进行Z轴堆叠扫描。轴突起始段长度测量基于锚蛋白G免疫荧光,通过覆盖整个神经元的Z轴堆叠最大强度投影图像完成。仅对起止点清晰可辨的轴突起始段结构,使用Fiji软件中的分段线工具进行分析。
¶ 突触密度定量
使用 油浸物镜和 变倍的空气扫描模块分析突触密度,对WT和突变体使用相同的成像参数。每个类器官采集3-6个感兴趣区域。使用内置分析软件(尼康系统)评估兴奋性突触前(SYN1)与突触后(PSD95)斑点的共定位情况,每批次对WT和突变体均采用一致的分析设置。将共定位斑点的突触密度标准化至WT组,以评估突变体组的变化。
¶ 蛋白质印迹法
类器官和组装体在含有蛋白酶和磷酸酶抑制剂(Thermo Fisher, A32953)的冰预冷RIPA裂解液(Thermo Fisher, 89901)中均质化后,于 离心20分钟。采用PierceTM BCA蛋白检测试剂盒(Thermo Fisher, 23225)测定蛋白浓度。蛋白样品在 Laemmli缓冲液( Tris-HCl pH 6.8、 SDS、 甘油、 溴酚蓝)中经 煮沸5分钟变性。取 样品上样于 SDS-PAGE凝胶,以80-120 V电压电泳后,在 条件下以 恒流转移2.5小时至PVDF膜(Thermo Fisher, PB9220)。膜在含 脱脂奶粉的TBST缓冲液(含 Tween 20的Tris缓冲盐溶液)中室温封闭1小时,随后与稀释于Intercept® T20(TBS)抗体稀释液(LI-COR Biosciences, 927-65001)的一抗于 孵育过夜。次日,膜经TBST洗涤 次(每次10分钟),室温下与二抗孵育1小时后再洗涤 次(每次10分钟)。采用Odyssey® CLx成像系统(LI-COR Biosciences)检测免疫反应条带,并通过Fiji软件分析。蛋白表达水平以 -actin为内参进行标准化比较。
¶ RNA提取、反转录和定量PCR分析
采用 RNeasy Mini 试剂盒(QIAGEN,#74104)从 hCO-hStrO 组装体中提取总 RNA,操作遵循制造商提供的方案。使用 NanoDrop 分光光度计(Thermo Fisher)评估 RNA 完整性与浓度。在最佳条件下使用 Maxima First Strand cDNA 合成试剂盒(Thermo Fisher,#K1672)进行反转录,以确保高效合成 cDNA。
采用PowerUp SYBR Green Master Mix(Thermo Fisher,货号#A25777)和基因特异性引物,在C1000 Touch PCR热循环仪(Bio-Rad)上进行定量PCR(qPCR)检测,操作遵循制造商推荐流程。热循环条件如下: 初始变性1分钟,随后进行45个循环( $9 5 ^ { \circ } \mathrm { C } 1 5 $ 秒, 秒)。SCN2A扩增使用内部SCN2A引物(正向:GAGACCATGTGGGACTGTATG;反向:AAGGCCAAGAAGAGGTTCAG)、密码子优化SCN2A引物(正向:GTGTTTTGCCTCTCCGTGTT;反向:ATTTCCGTCCAGGGAGTTGT)及总SCN2A引物(正向:GGATACATCTGTGTGAAGGC;反向:CTGTTCCTCATAGGCCAT)。
基因表达水平以GAPDH mRNA为内源性对照进行归一化,计算公式为( Ct)。
Ct值 Ct值Target Gene−Ct值Internal Control 相对表达量 = 2−ΔCt
所有反应均进行平行重复以确保可重复性,并使用Bio-Rad CFX Manager软件分析数据。
¶ 批量RNA测序与分析RNA提取和文库构建
从三个野生型(WT)细胞系(KOLF2.1J、B07、C03)、三个杂合(HET)SCN2A-C959X细胞系(A02、E04、F01)以及三个纯合(HOM)SCN2A-C959X细胞系(A03、D06、F03)衍生的5月龄hCO-hStrO组装体中提取RNA,共涉及两个独立批次的21个组装体(每种基因型7个)。使用NEBNext® Poly(A) mRNA磁性分离模块(New England Biolabs)从 总RNA中分离出多聚腺苷酸化(Poly(A)+)RNA。遵循xGen RNA文库制备试剂盒(Integrated DNA Technologies, IDT)制造商说明,在文库构建过程中直接使用 oligo dT磁珠进行RNA片段化与洗脱。制备的文库经混合后,在Illumina NovaSeq 系统上使用300循环25B流动槽进行测序,每个样本产生3000–3600万条双端读长。使用BCL2FASTQ软件(版本1.8.4)生成FASTQ文件用于下游分析。
¶ 预处理、比对与基因型验证
原始FASTQ文件使用fastp 进行处理,以去除接头序列并修剪低质量碱基(Phred分数 )。修剪后长度小于50 bp的读段被丢弃。剩余读段使用STAR比对工具(v2.7.10a)41以双通模式比对至GRCh38人类参考基因组(Ensembl 104版本),以提高剪接位点检测的准确性。为确认SCN2A基因中C959X突变的基因型,采用GATK单倍型识别器(v4.2.2.0)42进行联合基因分型43变异检测。该分析确保了样本能准确分为纯合突变(HOM)、杂合突变(HET)和野生型(WT)组。
¶ 基因定量与标准化
使用featureCounts(v1.6.1)44在双端、反链模式下进行基因组特征的读数分配。初步探索性分析采用DESeq2进行。
在 中用于评估样本聚类和整体一致性。原始计数矩阵经过过滤,保留至少在两个样本中拥有不少于5个读数的基因,随后通过计算偏差残差,在 RUVSeq (v1.28.0)47 中进行批次效应校正。数据标准化采用上四分位数法,通过 betweenLaneNormalization 函数实现。
¶ 差异表达与通路分析
对计数数据采用广义线性模型回归方法,将突变基因型作为协变量纳入,同时利用RUVSeq因子进行批次效应校正( )。采用edgeR(v3.36.0)48拟合准似然负二项模型进行差异表达分析,使用Benjamini-Hochberg方法进行多重假设检验校正以确定统计学显著性。将错误发现率FDR 的基因判定为差异表达基因。通过R语言(v4.3.2)中的clusterProfiler(v4.10.0)49工具,对差异表达基因进行KEGG、Reactome通路及基因本体富集分析,背景基因集包含经RUVSeq校正后检测到的全部基因。另采用Ingenuity Pathway Analysis(IPA)50对生物学通路及疾病关联性进行人工注释解读。
¶ 数据可视化
热图:使用edgeR提取批次效应校正的百万计数值,并通过R软件(v4.2.1)中的ComplexHeatmap包(v2.14.0)51绘制成热图。
火山图:差异表达结果使用EnhancedVolcano包(v1.16.0)52进行可视化。网络图与点图:通路富集结果通过Enrichplot(v1.22.0)和clusterprofiler(v4.10.0)展示以突出关键基因互作。孤独症相关基因中的枢纽基因通过Cytoscape(v3.10.2)的cytoHubba模块(v0.1)进行识别与可视化。
¶ 量化与统计分析
在进行任何参数检验之前,均使用GraphPad Prism对数据正态性和方差齐性进行检验。两组间比较采用双尾Student t检验(参数检验)或双尾Mann-Whitney U检验(非参数检验)。多重比较则根据情况选用单因素/双因素方差分析配合Tukey事后校正(参数检验),或Kruskal-Wallis检验配合Dunn多重比较校正(非参数检验)。仅当主分析显示统计学显著性时才进行事后检验。所有图表中的误差棒代表平均值±标准误。统计学显著性定义为p ,显著性水平标记如下: 标记为*, 标记为**, 标记为***, 标记为****。