¶ 塔顿-布朗-拉赫曼综合征相关的DNMT3A突变会解除对皮质中间神经元分化的抑制,从而破坏神经元网络功能
加雷斯·查普曼1#、朱莉安娜·德特曼1#、约翰·R·爱德华兹2、法伊扎·巴图尔1、詹姆斯·E·休特纳3、拉马钱德兰·普拉卡萨姆1、西德尼·R·克拉姆普1、海莉·杰特1、哈里森·W·加贝尔4、克里斯汀·L·克罗尔1*
1 华盛顿大学医学院发育生物学系,密苏里州圣路易斯市
2 华盛顿大学医学院肿瘤学系内科,密苏里州圣路易斯市,63110
3 细胞生物学与生理学系,华盛顿大学医学院,密苏里州圣路易斯市,63110
4 神经科学系,华盛顿大学医学院,圣路易斯,密苏里州 63110 # 同等贡献
- 通讯作者
¶ 摘要
DNMT3A的致病性突变会导致塔顿-布朗-拉赫曼综合征(TBRS),这是一种以包括大脑在内的多个组织的体细胞过度生长和智力障碍(OGID)为特征的疾病。在本研究中,我们利用新的人类多能干细胞模型,模拟了不同程度的TBRS相关DNMT3A功能缺失,对TBRS的病因进行了研究。我们发现,TBRS腹侧前脑内侧神经节隆起(MGE)样祖细胞存在谱系特异性过度生长,部分原因是PIK3/AKT/mTOR通路信号增强,而通过调节该通路可改善这种表型。相比之下,在MGE样祖细胞分化为γ-氨基丁酸能中间神经元的过程中,DNA甲基化水平降低会导致神经元和突触基因过早表达,引发神经元早熟。因此,TBRS的γ-氨基丁酸能神经元表现出过度活跃,足以改变神经元网络的发育和结构,这可能是导致TBRS患者常见的智力障碍和自闭症谱系障碍的原因之一。总之,这项研究阐明了DNMT3A介导的基因抑制在人类皮质发育中的新作用,确定了调控γ-氨基丁酸能神经元生成和神经元网络功能的关键需求。这些发现还为TBRS和其他OGIDs(包括PIK3CA相关过度生长综合征和韦弗综合征)的相关致病机制提供了证据,为未来研究确定治疗这些相关疾病的共同范式奠定了基础并提供了理论依据。
¶ 引言
过度生长和智力障碍疾病(OGIDs)是一组罕见的神经发育障碍(Atterton等人,2025年),通常由介导表观遗传调控的蛋白质或PI3K/AKT/mTOR信号级联组件中的突变引起(Tatton-Brown等人,2017年)。其中,新生DNA甲基转移酶DNMT3A的突变会导致Tatton-Brown-Rahman综合征(TBRS),这是一种涉及自闭症谱系障碍(ASD)、脑过度生长和智力障碍的神经发育障碍(Ostrowski和Tatton-Brown,1993年;Tatton-Brown等人,2018年;Lane等人,2020年;Thomas等人,2024年)。已发现多种与TBRS相关的突变,每种突变都会导致DNMT3A功能缺失的程度不同(Smith等人,2021年),其中大多数涉及DNMT3A功能域中的杂合错义突变,而其他类型的突变则较为罕见(Tatton-Brown等人,2018年)。
人类死后样本(Kang等人,2011年)和小鼠脑样本(Feng等人,2005年)的表达数据显示,DNMT3A的表达始于妊娠中后期,在小鼠神经发育早期取代DNMT3B成为主要的从头甲基转移酶(Watanabe等人,2006年),并在此过程中调控神经前体细胞的分化(Wu等人,2012年)。然而,尽管小鼠脑发育过程中存在DNMT3A,TBRS小鼠模型仍表现出类似TBRS的解剖学和行为缺陷,且没有脑过度生长的迹象(Christian等人,2020年;Smith等人,2021年;Beard等人,2023年)。此外,由于这些TBRS建模研究迄今为止主要集中在出生后发育阶段(Christian等人,2020年;Smith等人,2021年;Beard等人,2023年),因此评估与TBRS相关的DNMT3A功能缺失如何改变早期神经发育并促成TBRS的病因仍然至关重要。此外,尽管其他几种OGID疾病的病因在人类模型中也研究不足,但最近的证据表明,另一个与OGID相关的基因EZH2在抑制人类神经元分化中具有必要性(Ciceri等人,2024年),而来自小鼠模型的证据表明EZH2和DNMT3A的功能可能相互关联(Li等人,2022年)。因此,研究与TBRS相关的细胞病因,并验证是否存在趋同的分子机制导致其他形式的OGID,可为开发针对TBRS的特异性干预措施以及适用于所有OGID的共同干预措施奠定重要基础。
在此,我们构建了新的TBRS多能干细胞(hPSC)模型,并研究了DNMT3A功能缺失(LOF)突变如何改变人类皮质发育的关键方面。TBRS模型显示,内侧神经节隆起(MGE)样腹侧前脑神经祖细胞(V-NPCs)的增殖特异性增加,这与通过PIK3/AKT/mTOR通路的信号增强相关。在GABA能中间神经元发育的二维和三维模型中,TBRS模型还表现出神经发生和神经元成熟的增加,这与DNA甲基化降低导致的神经元基因表达提前有关。这导致GABA能神经元过度活跃,足以破坏神经元网络的同步性。总之,这项研究表明GABA能神经元对TBRS相关的DNMT3A突变特别敏感,并且还确定了与相关OGID基因失调相关的趋同机制。
¶ 结果
¶ TBRS模型显示V-NPC增殖和mTOR信号传导增强。
为了研究TBRS相关突变对神经元发育和功能的影响,我们建立了四组新的TBRS模型,涵盖不同程度的DNMT3A功能缺失(LOF),这与TBRS患者等位基因中观察到的DNMT3A功能缺失范围一致(Nguyen等人,2019年;Christian等人,2020年;Smith等人,2021年;Beard等人,2023年)。这些模型包括:一名携带p.R882H突变(882)患者的诱导多能干细胞(iPSC)模型,我们对这一反复出现的严重功能缺失(LOF)变体进行了校正,以生成同源对照(C-WT,图1a);携带p.P904L突变(904)敲入变体的人胚胎干细胞(hESC)模型(WT);以及在两个DNMT3A等位基因中均携带小缺失的hESC模型(KO,图1b)。我们还从一名存在一个DNMT3A等位基因全基因缺失的患者(Del1/2)中获得了另外两个iPSC模型,并将其与来自神经典型个体的两个不相关的性别匹配对照iPSC模型(Con1/2,图1c)配对。最后,我们建立了两个CRISPRi DNMT3A敲低模型(G1/G2)及其同源对照(KRAB)(图1d)。这些人多能干细胞模型(hPSCs)的总DNMT3A蛋白水平显示,正如预期的那样,882或904模型中几乎没有变化(图1e-f),但KO、Del1/2和G1/G2模型中DNMT3A蛋白显著减少(图1f-h)。
由于脑过度生长是TBRS临床表现的核心特征,我们首先评估了这些TBRS模型在具有背侧(D-,图1i)或腹侧(V-,图1j)端脑特征的神经祖细胞(NPCs)分化过程中是否表现出生长改变。初步观察显示,在882、904和Del1模型中,V-NPCs的神经球生长增加,而D-NPCs则无此现象(图1k-l,补充图S1a-d)。对Ki67+细胞的进一步量化证实,在这些TBRS模型中,V-NPCs的增殖特异性增加(图1m-o,补充图S1e-g),而对谱系特异性标志物(D-NPCs中的PAX6或V-NPCs中的NKX2.1)的评估显示,对照组和TBRS组NPCs之间无显著差异(补充图S1h-m)。在KO、Del2和CRISPRi(G1/G2)V-NPCs中的类似评估也显示V-NPCs的增殖特异性增加(图1p,补充图S2a-d),这证实了这是与TBRS相关的核心表型。
由于PI3K/AKT/mTOR通路基因的突变也会导致一种OGID疾病(PIK3CA相关过度生长综合征,PROS),且该通路是已明确的神经前体细胞(NPC)增殖调节因子(Romanyuk等人,2024年),我们接下来评估了TBRS的腹侧神经前体细胞(V-NPCs)中该通路的信号传导是否发生改变,结果发现AKT3、mTOR以及mTOR的靶标核糖体蛋白S6的表达和/或磷酸化水平在TBRS的V-NPCs中出现特异性且一致性的升高(图1q-r,补充图S3a-d)。这证实了PI3K/AKT信号传导增强与V-NPC增殖增加相关。我们进一步测试了降低mTOR活性是否能纠正TBRS相关的V-NPC增殖增加,结果表明雷帕霉素处理显著降低了TBRS模型中V-NPC的增殖(图1s,补充图S3e-f)。综上所述,这些结果表明V-NPCs对TBRS突变高度敏感,同时将DNMT3A功能缺失(LOF)与PROS潜在的信号传导异常联系起来,提示在遗传上不同的OGID疾病中,共同的分子机制是脑过度生长的基础。
¶ DNMT3A在腹侧神经祖细胞(V-NPC)特化过程中抑制神经元基因表达的起始。
为了明确NPC中TBRS突变所破坏的分子机制,我们分析了TBRS(882和904)与对照V-NPC之间的基因表达变化,发现不同TBRS模型既有不同的影响,也有共同的影响(图2a-b,补充数据1)。聚焦于在两种模型中同样失调的差异表达基因(DEGs)(共同DEGs,图2b,补充数据1),我们发现大多数在TBRS模型中上调(图2b,补充数据1),且总体而言,在882 V-NPC中的效应量大于904 V-NPC(补充图S4a,补充数据1)。虽然TBRS模型中上调的部分共同DEGs编码细胞增殖调节因子,这与我们观察到的V-NPC过度生长一致,但大多数是与神经元分化相关的基因(图2c,补充数据2),这表明DNMT3A还有第二个核心功能。通过评估882(严重LOF)、904(轻度LOF)、G2(轻度LOF)和G1(最轻度LOF)V-NPC中30个此类共同DEGs的表达,我们发现这些基因的表达增加与DNMT3A LOF的严重程度相关(图2d,补充图S4b),证实了DNMT3A在调节增殖基因和神经元基因表达中具有实质性作用。然而,尽管神经元基因表达有这种实质性增加,TBRS V-NPC仍保留祖细胞特性(补充图S4c,补充数据1),这表明仅DNMT3A LOF不足以启动神经元分化。相比之下,对882 D-NPC的平行转录组学评估显示,DNMT3A LOF突变对基因表达几乎没有影响,这与在TBRS D-NPC中未观察到细胞表型的结果一致(补充图S4d,补充数据1)。
接下来,我们通过鉴定882和904 V-NPC中DNA甲基化(mCG)的变化,研究了TBRS相关转录组失调的近端原因,发现882 V-NPC存在显著的整体和区域mCG降低(图2e;补充图S5a)。聚焦882模型,我们鉴定出差异甲基化区域(DMR),其中大多数在882 V-NPC中丢失了mCG(低甲基化DMR,补充数据3),这些位点通常与TBRS V-NPC中上调的突触和神经元基因相关(图2f-g,补充数据4)。对882和904 V-NPC中的mCG变化进行检查后,我们发现,在两个TBRS模型中存在的所有DMR的超集(所有DMR,图2h)以及882低甲基化DMR(图2h)中,882 V-NPC比904 V-NPC表现出更大的mCG丢失,这与这些模型中DNMT3A功能丧失的差异一致。当检查两个TBRS模型中持续低甲基化的DMR亚集时,我们发现两个模型的mCG水平都有类似的降低(图2h)。此外,这些共享DMR通常位于神经元基因的启动子处(补充图S5b)(图2i,补充数据4),其中许多也是共享差异表达基因(补充数据4)。对882 D-NPC的平行分析也揭示了mCG的实质性变化(补充数据3),但这些变化与实质性的转录组变化不相关。总之,这些结果表明,TBRS相关突变会破坏DNMT3A在V-NPC特化过程中抑制神经元基因表达起始的能力。
¶ 模拟腹侧前脑发育的TBRS类器官表现出神经发生增加
为了研究TBRS相关改变对神经祖细胞(NPC)发育的影响,我们分析了背侧模式(D-ORG,图3a)和腹侧模式(V-ORG,图3b)的前脑样类器官在发育过程中的变化。与我们在二维腹侧神经祖细胞(VNPC)诱导方案中观察到的增殖增加情况相比,我们发现TBRS(882、904和Del1)腹侧类器官中Ki67+增殖细胞比例同样增加(图3c,补充图S6a),而对腹侧神经祖细胞标志物DLX2和NKX2.1的评估显示,TBRS腹侧类器官的细胞命运决定未出现一致性改变(补充图S6b、c)。鉴于在TBRS腹侧神经祖细胞中观察到神经元基因表达增加,我们还检查了TBRS腹侧类器官中神经发生的改变,发现TUJ1免疫阳性区域显著增加(图3d、e),这并非由于SOX2+祖细胞的减少(图3d、f-g)。因此,为了评估TUJ1免疫阳性区域的增加是否由神经发生增强所致,我们还检查了促神经发生标志物ASCL1阳性细胞或未成熟γ-氨基丁酸能神经元标志物SST阳性细胞的比例,发现在TBRS腹侧类器官中ASCL1+细胞和SST+细胞均有所增加(图3h-i,补充图S6d-e),这证实了TBRS腹侧类器官中神经发生增强。
在D-ORGs中进行类似评估时,我们没有发现增殖改变的一致证据(补充图S7a),这再次与我们之前在二维模型中的观察结果一致。然而,我们确实观察到904个D-ORGs中SOX2+细胞的比例增加,而在所有TBRS D-ORGs中,TUJ1免疫阳性区域没有显著变化(补充图S7b-c)。最后,我们使用ASCL1、放射状胶质细胞标志物TBR2和早期出生的皮质谷氨酸能神经元标志物TBR1评估了神经发生的变化,发现在TBRS D-ORGs中,这些细胞群的比例均没有一致的差异(补充图S7d-f)。因此,与我们在NPC分化的二维模型中的发现一致,我们的类器官模型也表明,TBRS相关的DNMT3A功能丧失对D-NPC的发育影响甚微,但会在V-NPC发育过程中导致增殖和神经发生增加,这提示γ-氨基丁酸能神经元的产生和/或成熟增加。
¶ 抑制性H3K27me3可弥补DNA甲基化的严重缺失。
先前的研究还表明,另一个与OGID相关的基因EZH2在人类谷氨酸能神经元分化过程中对抑制神经元成熟具有作用(Gibson等人,2012年;TattonBrown等人,2013年)。由于这与我们关于DNMT3A功能丧失突变的发现相似,我们研究了在TBRS模型中,由EZH2沉积的组蛋白H3赖氨酸27三甲基化(H3K27me3)水平是否发生了改变。通过在882个D-NPC和V-NPC中识别H3K27me3富集差异位点(DKSs),我们发现TBRS模型中大多数位点的H3K27me3水平升高(高DKS,图4a-b,补充数据5)。有趣的是,虽然与882个低甲基化区域(hypo-DMRs)相交的D-NPC和V-NPC高DKSs与参与早期神经发育的基因相关(图4c-d,补充数据6),但其中很少是TBRS差异表达基因(DEGs)。因此,我们推测H3K27me3介导的抑制作用可能会补偿882个NPC中mCG的减少,潜在地防止因DNMT3A功能丧失导致的转录组失调,特别是在D-NPC中,因mCG低甲基化引起的转录组变化较轻微。
为了验证这一假设,我们在882个D-NPC中筛选出一组基因,这些基因既与低甲基化差异区域(hypo-DMRs)相关,又与高甲基化K27区域(hyper-DKSs)相关,但不属于TBRS差异表达基因(例如ESRG和ARX,图4e),并测试了破坏H3K27me3的沉积是否会改变它们的表达。在882个TBRS D-NPC中,无论是使用小干扰RNA(siRNA)敲低EZH2(图4f,补充图S8a),还是用选择性化学抑制剂(EZH2i)抑制EZH2的活性,都导致靶基因表达较对照组升高(图4g-h)。值得注意的是,在DNMT3A功能缺失程度较轻的D-NPC(904;补充图S8b)中未观察到这些效应,这表明在882模型中,更严重的mCG缺失可能触发了H3K27me3的代偿性增加。基于这些发现,我们还测试了EZH2过表达(OE)是否能逆转TBRS相关的基因表达升高,结果发现EZH2过表达部分挽救了TBRS相关的转录组失调(图4i-j)。综上,这些结果表明,抑制性的H3K27me3可代偿由严重mCG缺失引起的DNA甲基化降低,而增强的EZH2活性能够改善某些与TBRS相关的分子表型,这为TBRS与由EZH2突变引起的Weaver综合征之间存在机制联系提供了证据。
¶ TBRS突变会导致GABA能神经元过早成熟
接下来,我们研究了在TBRS V-NPCs中发现的神经元基因表达升高现象在其分化为未成熟的GABA能皮质中间神经元(V-INs;图5a)后是否持续存在。与我们对V-NPC的分析结果相似,我们发现882 V-INs中的转录组差异比904 V-INs更大(图5b,补充数据7),而在TBRS V-INs中上调的共有差异表达基因(shared-DEGs)再次富集于突触基因(图5c,补充数据8)。TBRS V-IN共有差异表达基因的效应量在882 V-INs中通常也比在904 V-INs中更大,42个与突触相关的共有差异表达基因便是例证(图5d,补充数据8)。我们还利用已发表的TBRS小鼠模型(p.R878H)成年皮质数据进行了比较基因集富集分析,该小鼠模型与882人类模型相似(Beard等人,2023年),结果发现神经元和突触基因存在共同上调(补充数据9)。总之,这些结果为突触基因失调作为与TBRS突变相关的核心表型提供了充分证据。
为了确定这些突触基因的持续上调是否由TBRS V-IN中持续的mCG缺失引起,我们检测了882和904个V-IN中的DNA甲基化情况,结果显示其mCG缺失程度比在V-NPC中观察到的要轻(补充图S9a-b)。然而,对TBRS V-IN中的差异甲基化区域(DMR)进行鉴定后发现了许多低甲基化DMR(补充数据10),其中904个V-IN的低甲基化DMR在882个V-IN中频繁被检测到,反之则不然(图5e,补充数据10)。聚焦于882和904个V-IN共有的低甲基化DMR(图5f,补充数据10),并将其与V-IN共有的差异表达基因(sharedDEGs)相关联后,再次发现了突触基因的富集(补充数据8)。我们还发现,尽管882和904的低甲基化DMR与突触基因的关联相似,但882的低甲基化DMR在发育过程中持续存在于相同位点,而904的低甲基化DMR的位置则更具时间特异性(图5f,补充数据10)。综上所述,这些发现表明,在TBRS GABA能神经元分化过程中,DNA甲基化的持续降低以及伴随的突触基因上调是一个持续存在的特征。
为了评估这些分子表型如何影响神经元成熟,我们接下来评估了TBRS GABA能神经元是否表现出形态改变,对第30天(D30)的对照组和TBRS(882、904和Del1)腹侧中间神经元(V-INs)进行了评分。该分析显示,TBRS腹侧中间神经元的分支增多,具体表现为次级神经突生成增加,但总神经突长度减少(图5g-i,补充图S10a),同时还发现TBRS腹侧中间神经元的胞体大小增大(补充图S10b)。我们还通过在第40天(D40)检查CALB1+和SST+腹侧中间神经元的生成情况,评估了DNMT3A功能缺失(LOF)是否同样会破坏皮质中间神经元的特性,结果发现TBRS腹侧中间神经元培养物中这两种标志物的免疫阳性比例均无显著差异(图5j-k,补充图S10c-f)。最后,我们评估了TBRS腹侧中间神经元的突触形成是否受到破坏,发现在第50天(D50)的TBRS腹侧中间神经元中,突触标志物(SYN1、PSD95和VGAT)的密度增加(图5l-p)。总之,这些结果证实,腹侧中间神经元分化过程中与TBRS相关的转录组失调也会导致腹侧中间神经元过早成熟。
在未成熟的谷氨酸能神经元(D-INs)中进行平行转录组分析时,我们发现TBRS(882和904)与对照D-INs之间存在显著的转录组差异(补充图S11a-b),其特征是人类多能干细胞(hPSCs)和D型神经前体细胞(D-NPCs)中正常表达的基因持续表达(补充图S11c-d,补充数据11-12)。我们通过绕过神经前体细胞(NPC)的特化过程来测试这种现象是否是发育改变的结果,而是通过诱导NGN2过表达来获得882和904谷氨酸能样神经元(iGluts,补充图S11e);这表明TBRS的D-INs和iGluts中存在类似的转录组失调(补充图S11f,补充数据11-12)。我们还发现,在882的D-INs和iGluts中存在类似的整体甲基化胞嘧啶(mCG)缺失,但904中没有(补充图S11g-j),同时在TBRS中识别出的差异甲基化区域(DMRs)显示D-INs和iGluts中存在类似的mCG缺失(补充图S11k;补充数据13)。D-INs中TBRS共有的低甲基化差异甲基化区域(hypo-DMRs)通常与神经元和突触基因相关(补充数据12),并且与这一观察结果一致,我们发现882的D型中间神经元(D-INs)和V型中间神经元(V-INs)中的差异甲基化区域(DMRs)显示出类似的甲基化胞嘧啶(mCG)缺失(补充图S11l,补充数据12-13)。综上所述,我们上述的研究表明,由于神经元和突触基因表达的持续失调,TBRS突变深刻地改变了γ-氨基丁酸能神经元的发育,而TBRS的谷氨酸能神经元则表现出明显的转录组变化,这在很大程度上与神经前体细胞(NPC)特化的改变和甲基化胞嘧啶(mCG)缺失无关。
¶ 与TBRS相关的GABA能神经元功能障碍会导致神经元网络超同步。
接下来,我们通过对在大鼠星形胶质细胞上单细胞培养并进一步成熟的谷氨酸能或γ-氨基丁酸能神经元(分别为成熟的D-INs或V-INs)进行膜片钳电生理实验,评估了TBRS突变的功能后果。初步评估显示,882个谷氨酸能神经元未出现明显的功能改变,这与D-IN分化过程中缺乏细胞表型的结果一致(补充图S12a-d,补充数据14)。相比之下,882个γ-氨基丁酸能神经元在受到刺激时表现出显著的过度活跃(图6a-b),而神经元健康状况未发生明显改变(图6c,补充图S13a-d,补充数据14)。这种过度活跃的特征是动作电位(AP)振幅增加和AP半峰宽减小(图6d-e)。我们还观察到882个γ-氨基丁酸能神经元中钠摄取增加、瞬时外向电流(TOC)增强以及对γ-氨基丁酸的反应性提高(图6f-h,补充图S13e)。当对904个谷氨酸能或γ-氨基丁酸能神经元的类似数据进行合并和分析时,我们同样发现了刺激后神经元过度活跃的证据(图6i中的示例,合并数据见图6j),而这两种神经元在其他神经元健康或活动指标上均未显示出显著变化(补充图S13f-k,补充数据14)。
接下来,我们使用低密度(LD)多电极阵列(MEA)来评估对照组或TBRS谷氨酸能神经元与γ-氨基丁酸能神经元共培养物中神经元网络活动的变化。该分析显示不同模型的结果存在差异,882培养物的活动增强(图7a-d;补充图S14a-b),而904培养物的趋势与这一发现不一致(补充图S14c-g)。因此,为了更清晰地研究这些表型,我们使用高密度(HD)MEA评估神经元网络活动,重点关注TBRSγ-氨基丁酸能神经元的功能障碍。在检查与对照iGluts共培养的TBRS或对照组第50天(D50)γ-氨基丁酸能神经元时,我们观察到平均放电率(MFR)没有差异,但表现出爆发行为的电极数量有所增加(图7e-g)。此外,在D82评估相同的培养物时,含有TBRSγ-氨基丁酸能神经元的共培养物的MFR升高,且含有882γ-氨基丁酸能神经元的培养物比含有904γ-氨基丁酸能神经元的培养物效果更显著(图7h-i)。这同时伴随着882和904模型的峰电位间期(ISI)缩短以及ISI变异系数(CoV)增加,其中含有882γ-氨基丁酸能神经元的培养物再次显示出更显著的效果(补充图S15a-b)。
鉴于γ-氨基丁酸能神经元在同步神经元活动中的重要性(Makinen等人,2018年;Kirmse和Zhang,2022年),我们进一步研究了这些D82共培养物中神经元爆发和神经元网络活动的变化。该分析显示,在含有TBRSγ-氨基丁酸能神经元的培养物中,爆发神经元的动作电位(AP)高度降低(补充图S15ce),且每个爆发中的尖峰数量增加(图7j)。此外,含有882γ-氨基丁酸能神经元的培养物不仅爆发频率增加(补充图S15f),爆发中尖峰的比例也有所上升,同时爆发内的尖峰间期(ISI)缩短(图7k,补充图S15g)。最后,虽然我们未观察到与两种TBRS模型相关的神经元网络活动频率发生改变(图7l),但TBRSγ-氨基丁酸能神经元导致网络爆发持续时间缩短(图7m),同时网络爆发内的尖峰数量和比例增加(图7n,补充图S15h)。这些变化还伴随着网络爆发内尖峰间期的缩短以及尖峰间期变异系数(CoV)的增加(图7o,补充图S15i)。总之,这些数据表明,882和904两种与TBRS相关的γ-氨基丁酸能神经元功能障碍均足以引发神经元网络的过度同步。
¶ 讨论
在本研究中,我们构建了新的TBRS人类模型,并利用这些模型表征了与TBRS相关的神经发育和神经元功能改变,发现γ-氨基丁酸能神经元的发育对致病性DNMT3A突变具有选择性敏感性。我们确定了DNMT3A在γ-氨基丁酸能神经发育过程中对抑制增殖和神经发生的关键作用。我们证明,在TBRS模型中,DNA甲基化的破坏会导致γ-氨基丁酸能神经元分化过程中神经元和突触基因的过早表达,从而驱动γ-氨基丁酸能神经发生,并最终导致神经元过度活跃。这种γ-氨基丁酸能神经元的过度活跃足以破坏神经元网络的形成,导致神经元网络的过度同步。最后,我们强调了TBRS与其他由其他表观遗传修饰因子以及PIK3/AKT/mTOR信号轴突变引起的OGID之间的机制联系。总之,这些发现揭示了DNMT3A可能在TBRS发病机制中发挥的新的发育和谱系特异性作用。
值得注意的是,在我们的模型中,TBRS相关LOF的差异程度与表型严重程度相关,这让人联想到882和904同源TBRS小鼠模型的研究结果(Smith等人,2021年;Beard等人,2023年)。然而,迄今为止的小鼠TBRS研究主要集中在成年动物有丝分裂后神经元中由DNMT3A催化的非CG甲基化(Christian等人,2020年;Beard等人,2023年),而我们发现,在人类TBRS模型中,GABA能神经元的异常发育是神经元网络过度活跃和同步性过高的基础。同样,在人类运动神经元(hMN)发育过程中敲除DNMT3A纯合体会改变hMN功能,导致过度活跃;但这伴随着神经发生和突触生成受损(Ziller等人,2018年),与我们的研究结果相反。我们的研究结果反而表明,DNMT3A在抑制皮质神经元成熟方面发挥作用,这揭示了TBRS突变导致DNMT3A功能破坏所产生的不同细胞类型特异性后果。
我们还在所有TBRS模型中发现了GABA能神经元特化过程中的过度增殖,这可能是导致TBRS患者脑过度生长的原因之一。鉴于在TBRS小鼠模型中尚未有脑过度生长的相关报道(Christian等人,2020;Beard等人,2023),我们的研究表明这可能是人类特有的现象,这与其他几种神经发育障碍的研究结果一致(Ernst,2016;Marchetto等人,2017;Wang等人,2020;Connacher等人,2022)。我们的结果还支持TBRS和PROS之间存在功能相互作用,TBRS模型显示出PIK3/AKT/mTOR信号通路活性增强,这与该通路组件的功能获得性突变会导致相关脑过度生长疾病PROS的发现一致(Mirzaa等人,2016)。尽管对PROS相关突变的研究仍不够深入(Mirzaa等人,2016;Dobyns和Mirzaa,2019),但已知该信号轴在癌症中会促进细胞过度增殖(Samuels等人,2005;Wang等人,2017;Chen等人,2022),因此PIK3/AKT/mTOR信号通路的药理学调节剂应用广泛。我们的研究强调了雷帕霉素在调节过度增殖中的作用,而先前的研究已使用该信号轴的其他调节剂来纠正与TSC缺陷相关的神经元网络异常(Alsaqati等人,2020),TSC缺陷同样会破坏GABA能神经元的分化(Fu等人,2012)和功能(Bassetti等人,2021)。因此,正如我们的数据所表明的,如果PIK3/AKT/mTOR信号通路异常是所有脑过度生长障碍(OGIDs)的共同特征,那么这可能为开发协同干预手段以治疗多种遗传背景不同的脑过度生长障碍提供机会。
我们的研究结果还支持EZH2和DNMT3A之间存在抑制人类γ-氨基丁酸能神经元分化的机制性相互作用,这表明一种共同机制可能是TBRS和韦弗综合征(一种由EZH2突变引起的OGID)的病因基础(Tatton-Brown等人,2013年;Cohen等人,2016年;Tatton-Brown等人,2017年)。最近的研究表明,EZH2在抑制谷氨酸能神经元成熟中发挥作用(Ciceri等人,2024年),这提示其他与OGID相关的基因在调节神经元成熟中具有关键作用。同样,在致病性的人类模型中,神经元成熟也受到了破坏。
MECP2突变可模拟雷特综合征(Landucci等人,2018年),这种突变会破坏MECP2对DNMT3A沉积的DNA甲基化的读取(Clemens等人,2020年;Sandweiss等人,2020年;Pantier等人,2024年)。此外,在雷特综合征小鼠模型中,特异性恢复GABA能神经元中的MECP2表达,可挽救多种与雷特综合征相关的表型(Ure等人,2016年)。结合我们的研究结果,这些发现证实了GABA能神经元对神经发育障碍(NDD)相关扰动具有选择性敏感性,而这些扰动会破坏神经元成熟的表观遗传抑制。
我们的高密度微电极阵列(HD-MEA)结果进一步表明,γ-氨基丁酸能神经元功能障碍足以导致结节性硬化症(TBRS)中兴奋性和抑制性信号失衡,而这种兴奋/抑制(E/I)失衡是被认为是神经发育障碍(NDD)发病机制的常见机制(Gatto和Broadie,2010;Uzunova等人,2016;Canitano和Pallagrosi,2017;Markicevic等人,2020;Pietropaolo和Provenzano,2022)。尽管对神经发育障碍模型的微电极阵列(MEA)评估正变得越来越普遍,但低密度微电极阵列(LD-MEA)研究得出的结论往往受到数据量有限的限制,这常常导致神经发育障碍研究内部和跨研究之间的结果不一致(Nageshappa等人,2016;Marchetto等人,2017;Amatya等人,2019;Deneault等人,2019a;Deneault等人,2019b;Frega等人,2019;Alsaqati等人,2020;Graef等人,2020;Chapman等人,2022;DeRosa等人,2022;Rylaarsdam等人,2024)。因此,尽管在雷特综合征细胞模型中进行的低密度微电极阵列评估揭示了与我们的结节性硬化症模型一些表型相似性(Mok等人,2022),但我们的研究强调了使用高密度微电极阵列来解决由低密度微电极阵列实验中有限数据导致的不一致性的重要性。最终,我们在这里提出的使用高密度微电极阵列的范式提供了一个高质量的工作流程,用于评估γ-氨基丁酸能神经元功能改变在破坏神经元网络发育中的作用,这一流程可应用于未来对寡糖基转移酶缺陷(OGIDs)和其他神经发育障碍的研究中。
总之,我们的研究表明,与TBRS相关的GABA能神经元功能障碍足以破坏神经元网络的发育,这可能是导致TBRS患者中常见的自闭症谱系障碍(ASD)和智力障碍(ID)的病因之一。此外,我们关于不同OGIDs之间分子和表型关系的研究结果,凸显了可能是它们共同临床表现基础的趋同机制。总之,这些结果表明,未来针对TBRS开发干预措施的努力可能在更广泛的OGIDs中具有适用性,同时提供了关于敏感细胞类型和发育时期的重要信息,这将有助于此类干预措施的开发。
¶ 方法
¶ 人多能干细胞模型的生成与培养
人多能干细胞(hPSCs)相关研究是按照华盛顿大学胚胎干细胞研究监督委员会(ESCRO)的#12-002号协议进行的。DNMT3A R882H诱导多能干细胞(iPSCs)是从一名携带杂合子p.R882H(c.2922G>A)突变的男性患者身上重编程而来,并采用CRISPR-Cas9技术进行了校正。对R882H诱导多能干细胞系及其经校正后的对照(C-WT)诱导多能干细胞系均进行了全基因组测序,汇总数据见补充数据16,完整数据可从dbGaP(PHS000159)获取。通过将诱导多能干细胞注射到小鼠脂肪垫中评估畸胎瘤形成情况,证实了R882H和C-WT诱导多能干细胞均能分化产生三个原始胚层。
Del1和Del2诱导多能干细胞模型同样源自一名男性患者,该患者携带包含整个DNMT3A基因的135kb杂合缺失。相关对照(Con1/2)购自富士胶片细胞动力学公司(CW20110和CW20098),为性别匹配的无血缘关系对照,无神经发育障碍病史,包括自闭症谱系障碍和癫痫。通过CRISPR-Cas9技术将杂合的P904L(c.2711C>T)突变导入H1人胚胎干细胞(野生型),构建了该突变模型;采用相同方法,在cDNA的c.2711位置附近,使DNMT3A基因的两个等位基因均发生小片段(<10bp)缺失,构建了纯合模型(敲除型)。
通过用单个载体(Lenti-(BB)-EF1a-KRAB-dCas9-P2A-BlastR)转导H1人胚胎干细胞(hESCs),构建了DNMT3A CRISPRi细胞系,该载体携带dCas9-KRAB和每个向导RNA(gRNA)的表达盒。来自Dolcetto文库(Sanson等人,2018年)的DNMT3A向导RNA(补充数据17)被克隆到Lenti-(BB)-EF1a-KRAB-dCas9-P2A-BlastR中。用于产生诱导性谷氨酸能样神经元(iGluts)的细胞系,是通过用携带pLVX-UbC-rtTA Ngn2:2A:EGFP的慢病毒转导细胞系而生成的。关于慢病毒生产和稳定细胞系生成的详细信息,请参见补充材料。
所有人类多能干细胞模型均在无饲养层条件下,于StemFlex培养基中的玻连蛋白上进行维持,并在所有实验过程中置于含5%二氧化碳、37°C的培养箱中培养。所有人类多能干细胞系的实验均在第20-50代之间进行,且各细胞系定期检测,结果均为支原体污染阴性。
¶ 皮质γ-氨基丁酸能神经元和谷氨酸能神经元发育的建模
皮质γ-氨基丁酸能神经元发育的建模按照先前描述的方法进行(Chapman等人,2024年),具体是将人多能干细胞指定为具有腹侧端脑祖细胞(V-NPCs)特征的内侧神经节隆起(MGE)样祖细胞,并将V-NPCs分化为未成熟的皮质γ-氨基丁酸能中间神经元(V-INs)。在分化的第15天收集细胞作为V-NPCs,在分化的第35天收集细胞作为V-INs。为进行如下所述的功能评估,对V-INs进行了进一步成熟处理。详细信息请参见补充材料。
皮质谷氨酸能神经元发育的建模按照先前描述的方法进行(Chapman等人,2022年),并稍作修改:将人多能干细胞(hPSCs)指定为具有背侧端脑祖细胞特征的脑室下区样祖细胞(D-NPCs),并将这些D-NPCs分化为未成熟的谷氨酸能神经元(D-INs)。在分化的第20天收集细胞作为D-NPCs,在分化的第40天收集细胞作为D-INs。如下所述,对D-INs进行进一步成熟以用于功能评估。详细信息请参见补充材料。
诱导的谷氨酸能样神经元按照先前描述的方法(Schafer等人,2019年)通过诱导Neurogenin-2(NGN2)的过表达来生成。细胞在分化14天后被视为诱导性谷氨酸能神经元(iGluts)。详细信息请参见补充材料。
具有背侧(D)或腹侧(V)端脑特征的类器官(Orgs)是通过将在D-NPC或V-NPC规格确定过程中生成的模式化神经球包埋在Matrigel®中而产生的。然后,D型和V型类器官在轨道摇床上维持培养,进行总共30天的分化;详细信息请参见补充材料。收集类器官,用4%多聚甲醛在4℃下固定过夜,随后在30%(v/v)蔗糖溶液中于4℃下孵育过夜。将类器官包埋在1:1比例的O.C.T复合物和30%蔗糖中,然后在低温恒冷切片机上切成8微米的切片。
¶ 细胞表型分析
通过蛋白质印迹法,利用从多能干细胞中分离出的蛋白质,评估了TBRS模型中DNMT3A表达的变化。在分化12天后(D12),对D-NPCs和V-NPCs中的神经球生长情况进行了定量分析,并在D-NPC或V-NPC定型完成4天后,通过免疫细胞化学法评估了增殖和谱系标志物表达的变化。分化30天后,在D-Orgs和V-Orgs中对类器官大小、增殖和谱系标志物表达进行了类似的定量分析。还对定型完成后分离的D-NPC和V-NPC样本进行了RT-qPCR,以评估特定基因表达的变化。
通过蛋白质印迹法利用V-NPC样本评估PIK3/AKT/mTOR信号通路的变化,在V-NPC分化完成后,将雷帕霉素(5 nM)应用于V-NPCs,持续4天,以评估mTOR抑制后增殖的变化。同样地,在分化完成后,通过RT-qPCR评估EZH2抑制的效果,实验中D-NPCs分别用EZH2抑制剂(GSK343,4 µM)与二甲基亚砜处理,或用靶向EZH2的小干扰RNA与针对GFP的小干扰RNA处理,持续4天。
通过免疫细胞化学方法,在第30天对D型类器官(D-Orgs)和V型类器官(V-Orgs)中神经元分化标志物进行检测,以此评估TBRS相关的神经元分化改变,同时在第40天对V型诱导神经元(VINs)进行类似的定量分析。在第30天对V型诱导神经元(V-INs)的神经元形态进行评估,并在V型诱导神经元与大鼠星形胶质细胞共培养20天后,于第50天对其突触生成情况进行评估。有关上述方法的更多信息,请参见补充材料。
¶ 测序
如前所述(Hamagami等人,2023年;Chapman等人,2024年),对第15天的882、904、C-WT和WT V-NPCs进行了RNA测序(RNA-seq)和全基因组亚硫酸氢盐测序(WGBS),并对第20天的882和C-WT D-NPCs进行了类似分析。如前所述(Chapman等人,2024年),对第15天的882、904、C-WT和WT V-NPCs进行了组蛋白H3赖氨酸27三甲基化的CUT&Tag实验。分别在第40天和第35天对882、904、C-WT和WT的D-INs和V-INs进行了RNA-seq和WGBS,并在第14天对882、904、C-WT和WT iGluts进行了类似分析。有关CUT&Tag、RNA-seq和WGBS方法的详细信息,请参见补充材料。
¶ 功能表型分析
在电生理学方面,将D28 D-INs和D23 V-INs接种在大鼠皮质星形胶质细胞上,培养至第60天,然后按照先前描述的方法进行电生理学测量(Meganathan等人,2017年)。使用非参数秩和检验对结果进行分析,将TBRS模型与匹配的同基因对照进行比较,原始结果见补充数据。
所有低密度多电极阵列(MEA)实验均使用CytoView MEA 48孔板进行,采用成熟的D-INs和V-INs共培养物(比例为70:30)。使用Axion Biosystems公司的硬件(Maestro Edge)和软件(AxIS 1.5.2)每5天记录一次电生理活动,并使用内置的Axion神经指标分析工具进行分析。
所有高密度MEA实验均使用CorePlate™ 6W 38/60 MEA进行,采用V-INs和iGluts的共培养物(比例为30:70)以及大鼠星形胶质细胞。在V-IN分化的第50天和第82天,使用HyperCAM Alpha系统(3Brain)及配套的BrainWave5软件记录电生理活动,并使用BrainWave5软件进行主要数据分析。有关TBRS模型的电生理和MEA表征的详细信息,请参见补充材料。
¶ 量化与统计分析
在适当情况下,使用GraphPad Prism 9版本(GraphPad Software公司;美国加利福尼亚州拉霍亚,可从www.graphpad.com获取)和/或RStudio 3.5.1版本(RStudio:R语言的集成开发环境;美国马萨诸塞州波士顿,可从www.rstudio.org获取)进行统计分析。在进行统计分析前,所有技术重复均取平均值,每项数据分析所使用的统计检验在图注中或特定分析范式的详细方法部分有详细说明。每个时间点或生物学条件至少使用3次独立分化实验,每个样本所用的分化实验次数在图注中以n表示。图中的结果以组平均值±标准误(SE)呈现,除非图注中另有说明,否则表示用于分析的每个生物学重复。统计显著性标注如下:n.s.表示无显著性;∗表示p < 0.05;∗∗表示p < 0.01;∗∗∗表示p < 0.001;∗∗∗∗表示p < 0.0001,除非图注中另有说明。
¶ 利益冲突
作者声明不存在利益冲突。
¶ 致谢
本研究得到以下机构的支持:美国国立卫生研究院(NIH)授予KLK的R01NS114551、R01MH124808、R01HD110556和U01HG007530(NIH共同基金/国家神经疾病和中风研究所未确诊疾病网络试点基因研究子奖项);NIH授予Joseph Dougherty和Christina Gurnett的P50HD103525(KLK是华盛顿大学智力和发育障碍研究中心人类细胞模型单元的项目负责人,该项目的研究经费来自此项资助)。支持本研究的基金会包括Engelhart家族基金会、西蒙斯基金会、M-CM网络,以及华盛顿大学希望中心、再生医学中心和临床与转化科学研究所授予KLK的试点奖项。感谢华盛顿大学医学院麦克唐奈基因组研究所的基因组技术获取中心提供的基因组学服务支持。该中心部分得到美国国立卫生研究院(NIH)下属国家研究资源中心(NCRR)及NIH医学研究路线图给予Siteman癌症中心的NCI癌症中心支持基金(编号P30 CA91842)的资助。本出版物仅代表作者的观点,不一定反映NCRR或NIH的官方意见。我们也感谢圣路易斯华盛顿大学的基因组工程与干细胞中心(GESC@MGI)提供的细胞系工程服务。本研究中使用的DNMT3A R882H诱导多能干细胞系和Del1/2诱导多能干细胞系由华盛顿大学Timothy J. Ley实验室的Daniel George和Christopher A. Miller博士开发并鉴定,研究得到NIH CA197161基金的支持。
¶ 作者贡献
G.C.和J.D.进行了分化和MEA评估,并分析了所有结果。G.C.和J.R.E.进行了计算和生物信息学分析。J.E.H和J.D.进行了电生理评估及相关分析。G.C.和S.C.进行了qPCR及相关分析。G.C.、F.B.、R.P.和H.J.进行了免疫细胞化学评估,G.C.和F.B.进行了蛋白质印迹分析。H.W.G.和K.L.K.监督了这项工作。G.C.撰写了手稿,所有作者都参与了手稿的审阅和编辑。
¶ 参考文献
Alsaqati M、Heine VM、Harwood AJ。通过药物干预恢复源自携带TSC2突变的自闭症谱系障碍患者诱导多能干细胞的神经元网络连接缺陷。《分子自闭症》。2020;11:80。
Amatya DN、Linker SB、Mendes APD、Santos R、Erikson G、Shokhirev MN、Zhou Y、Sharpee T、Gage FH、Marchetto MC 等人。自闭症谱系障碍患者神经元分化过程中动态电复杂性降低。《干细胞报告》。2019;13:474-484。
Atterton C、Trew I、Cale JM、Aung-Htut MT、Grens K、Kiernan J、Delagrammatikas CG、Piper M。过度生长-智力障碍疾病:生物学、患者权益倡导和创新疗法的进展。《疾病模型与机制》。2025;18。
Bassetti D, Luhmann HJ, Kirischuk S. TSC基因突变对神经发育和突触传递的影响。国际分子科学杂志。2021;22.
Beard DC、Zhang X、Wu DY、Martin JR、Erickson A、Boua JV、Hamagami N、Swift RG、McCullough KB、Ge X等。DNMT3A中不同的疾病突变导致一系列行为、表观遗传和转录缺陷。《细胞报告》。2023;42:113411。
Canitano R, Pallagrosi M. 自闭症谱系障碍与精神分裂症谱系障碍:兴奋/抑制失衡及发展轨迹。《精神病学前沿》。2017;8:69.
查普曼 G、阿尔萨卡蒂 M、伦恩 S、辛格 T、林登 SC、林登 DEJ、范登布里 MBM、齐勒 M、欧文 MJ、霍尔 J 等。利用诱导多能干细胞研究 1q21.1 缺失和重复综合征中的人类神经元表型。《分子精神病学》。2022;27:819–830。
查普曼 G、德特曼 J、耶特 H、考希克 K、普拉卡萨姆 R、克罗尔 KL。定义控制人类皮质中间神经元发育的顺式调控元件和转录因子。《交叉科学》。2024;27:109967。
陈W、戴G、钱Y、文L、何X、刘H、高Y、唐X、董B。PIK3CA突变通过PI3K-MEK/PDK1-GPT2通路影响结直肠癌细胞的增殖。《肿瘤学报告》。2022;47。
Christian DL、Wu DY、Martin JR、Moore JR、Liu YR、Clemens AW、Nettles SA、Kirkland NM、Papouin T、Hill CA 等。DNMT3A 单倍体功能不足导致神经发育障碍中共有的行为缺陷和全基因组表观调控异常。《细胞报告》。2020;33:108416。
Ciceri G、Baggiolini A、Cho HS、Kshirsagar M、Benito-Kwiecinski S、Walsh RM、Aromolaran KA、Gonzalez-Hernandez AJ、Munguba H、Koo SY 等人。一种表观遗传屏障决定了人类神经元成熟的时间。《自然》。2024;626:881-890。
克莱门斯 AW、吴 DY、摩尔 JR、克里斯蒂安 DL、赵 G、加贝尔 HW。MeCP2 通过染色体拓扑相关 DNA 甲基化抑制增强子。《分子细胞》。2020;77:279–293.e278.
科恩AS、亚普DB、刘易斯ME、千羽和、拉莫斯-阿罗约MA、特卡琴科N、米兰V、弗拉丁M、麦金农ML、汤森德KN等。 Weaver综合征相关EZH2蛋白变体在体外显示出组蛋白甲基转移酶功能受损。《人类突变》。2016;37:301-307。
Connacher R、Williams M、Prem S、Yeung PL、Matteson P、Mehta M、Markov A、Peng C、Zhou X、McDermott CR 等人。特发性和 16p11.2 拷贝数变异缺失患者的自闭症神经祖细胞均表现出增殖和有丝分裂反应失调。《干细胞报告》。2022;17:1786。
Deneault E、Faheem M、White SH、Rodrigues DC、Sun S、Wei W、Piekna A、Thompson T、Howe JL、Chalil L等人。来自自闭症患者的CNTN5(+/-)或EHMT2(+/-)人类诱导多能干细胞衍生神经元会形成过度活跃的神经元网络。《eLife》。2019a;8.
Deneault E、White SH、Rodrigues DC、Ross PJ、Faheem M、Zaslavsky K、Wang Z、Alexandrova R、Pellecchia G、Wei W 等。通过基因编辑完全破坏自闭症易感基因主要降低了同基因人类神经元的功能连接性。《干细胞报告》。2019b;12:427-429。
DeRosa BA、Hokayem JE、Artimovich E、Garcia-Serje C、Phillips AW、Van Booven D、Nestor JE、Wang L、Cuccaro ML、Vance JM 等。作者更正:通过患者来源神经元的时程转录组分析揭示特发性自闭症的 convergent 通路。《科学报告》。2022;12:3445。
Dobyns WB, Mirzaa GM. 与PI3K-AKT-MTOR通路核心成分(PIK3CA、PIK3R2、AKT3和MTOR)突变相关的巨脑症综合征。《美国医学遗传学杂志C辑:医学遗传学研讨会》。2019;181:582-590.
恩斯特·C. 增殖和分化缺陷是神经发育障碍的主要汇合点。《神经科学趋势》。2016;39:290-299。
冯J、常H、李E、范G。中枢神经系统中从头DNA甲基转移酶Dnmt3a和Dnmt3b的动态表达。《神经科学研究杂志》。2005;79:734-746。
Frega M、Linda K、Keller JM、Gumus-Akay G、Mossink B、van Rhijn JR、Negwer M、Klein Gunnewiek T、Foreman K、Kompier N等。由增强的NMDAR信号介导的Kleefstra综合征模型中的神经网络功能障碍。《自然·通讯》,2019;10:4928。
傅聪,考森,克林克斯凯尔斯,布鲁斯,温岑伯格,埃斯·KC。Tsc1基因调控γ-氨基丁酸能中间神经元的发育和功能。《大脑皮层》。2012;22:2111-2119。
加托CL,布罗迪K。神经发育障碍模型中平衡兴奋性和抑制性突触发生的遗传控制。《突触神经科学前沿》。2010;2:4.
吉布森WT、胡德RL、詹SH、布尔曼DE、费耶斯AP、摩尔R、芒格尔AJ、埃杜克斯P、巴布-希尔吉R、安J等。EZH2基因突变导致韦弗综合征。《美国人类遗传学杂志》。2012;90:110-118。
Graef JD、Wu H、Ng C、Sun C、Villegas V、Qadir D、Jesseman K、Warren ST、Jaenisch R、Cacace A等。部分FMRP表达足以使脆性X神经元的神经元过度活动正常化。《欧洲神经科学杂志》。2020;51:2143-2157。
滨上N、吴DY、克莱门斯AW、内特尔斯SA、李A、加贝尔HW。NSD1通过沉积组蛋白H3赖氨酸36二甲基化来调控神经元中非CG DNA甲基化模式。《分子细胞》。2023;83:1412–1428.e1417。
Kang HJ、Kawasawa YI、Cheng F、Zhu Y、Xu X、Li M、Sousa AM、Pletikos M、Meyer KA、Sedmak G等。人类大脑的时空转录组。《自然》。2011;478:483-489。
Kirmse K, Zhang C. 发育中皮质网络动态中GABA能信号的原理。《细胞报告》。2022;38:110568.
Landucci E、Brindisi M、Bianciardi L、Catania LM、Daga S、Croci S、Frullanti E、Fallerini C、Butini S、Brogi S等。iPSC衍生神经元的分析显示,雷特综合征中存在GABA能回路破坏和乙酰化α-微管蛋白缺陷,而iHDAC6治疗后这些情况有所改善。《实验细胞研究》。2018;368:225-235。
莱恩C、塔顿-布朗K、弗里思M。塔顿-布朗-拉赫曼综合征:认知和行为表型。《发育医学与儿童神经病学》。2020;62:993-998。
李J、平托-杜阿尔特A、赞德M、科沃MS、赖CY、奥斯丁J、方L、罗C、卢塞罗JD、戈麦斯-卡斯塔农R等。兴奋性神经元中Dnmt3a敲除会损害出生后突触成熟并增加抑制性组蛋白修饰H3K27me3。《eLife》。2022;11。
Makinen ME、Yla-Outinen L、Narkilahti S。γ-氨基丁酸(GABA)和缝隙连接在人多能干细胞衍生神经网络同步活动发展中的作用。《细胞神经科学前沿》。2018;12:56。
Marchetto MC、Belinson H、Tian Y、Freitas BC、Fu C、Vadodaria K、Beltrao-Braga P、Trujillo CA、Mendes APD、Padmanabhan K等人。特发性自闭症患者来源的神经细胞增殖及网络改变。《分子精神病学》。2017;22:820-835。
Markicevic M、Fulcher BD、Lewis C、Helmchen F、Rudin M、Zerbi V、Wenderoth N. 皮质兴奋-抑制失衡会导致异常的脑网络动态,这在神经发育障碍中可见。《大脑皮层》. 2020;30:4922-4937.
Meganathan K、Lewis EMA、Gontarz P、Liu S、Stanley EG、Elefanty AG、Huettner JE、Zhang B、Kroll KL。从人类胚胎干细胞中指定皮质中间神经元的调控网络揭示了CHD2在中间神经元发育中的作用。《美国国家科学院院刊》。2017;114:E11180–E11189。
Mirzaa G、Timms AE、Conti V、Boyle EA、Girisha KM、Martin B、Kircher M、Olds C、Juusola J、Collins S等人。PIK3CA相关发育障碍表现出不同类别的突变,具有不同的表达和组织分布。《JCI洞察》。2016;1.
Mok RSF、Zhang W、Sheikh TI、Pradeepan K、Fernandes IR、DeJong LC、Benigno G、Hildebrandt MR、Mufteev M、Rodrigues DC等。具有雷特综合征相关MECP2突变的人干细胞源性兴奋性神经元中神经元和网络表型的广泛谱。《转化精神病学》。2022;12:450。
Nageshappa S、Carromeu C、Trujillo CA、Mesci P、Espuny-Camacho I、Pasciuto E、Vanderhaeghen P、Verfaillie CM、Raitano S、Kumar A等人。人类MECP2重复模型中神经元网络的改变及挽救。《分子精神病学》。2016;21:178-188。
Nguyen TV、Yao S、Wang Y、Rolfe A、Selvaraj A、Darman R、Ke J、Warmuth M、Smith PG、Larsen NA等。R882H DNMT3A热点突变可稳定大型DNMT3A寡聚体的形成,且其DNA甲基转移酶活性较低。《生物化学杂志》。2019;294:16966-16977。
Ostrowski PJ, Tatton-Brown K. Tatton-Brown-Rahman综合征。收录于:Adam MP、Feldman J、Mirzaa GM、Pagon RA、Wallace SE、Bean LJH、Gripp KW、Amemiya A主编。《GeneReviews®》。西雅图(华盛顿州):华盛顿大学;1993年。
Pantier R、Brown M、Han S、Paton K、Meek S、Montavon T、Shukeir N、McHugh T、Kelly DA、Hochepied T 等人。MeCP2 不依赖相分离和异染色质组织结合甲基化 DNA。《自然·通讯》。2024;15:3880。
彼得罗保罗·S、普罗文扎诺·G。社论:针对神经发育障碍和自闭症谱系障碍中的兴奋-抑制失衡。《神经科学前沿》。2022;16:968115。
Romanyuk N、Sintakova K、Arzhanov I、Horak M、Gandhi C、Jhanwar-Uniyal M、Jendelova P。mTOR通路抑制改变神经干细胞的增殖和分化。《细胞神经科学前沿》。2024;18:1298182。
Rylaarsdam L、Rakotomamonjy J、Pope E、Guemez-Gamboa A。iPSC衍生的PACS1综合征模型揭示神经元活动的转录和功能缺陷。《自然·通讯》。2024;15:827。
塞缪尔斯 Y、迪亚兹 LA Jr、施密特-基特勒 O、康明斯 JM、德龙 L、张 I、拉戈 C、胡索 DL、伦高尔 C、金兹勒 KW 等。突变型 PIK3CA 促进人类癌细胞的生长和侵袭。《癌细胞》。2005;7:561-573。
Sandweiss AJ、Brandt VL、Zoghbi HY。对雷特综合征和MECP2重复综合征理解的进展:未来治疗的前景。《柳叶刀·神经病学》。2020;19:689-698。
Sanson KR、Hanna RE、Hegde M、Donovan KF、Strand C、Sullender ME、Vaimberg EW、Goodale A、Root DE、Piccioni F 等。用于多模态 CRISPR-Cas9 遗传筛选的优化文库。《自然·通讯》。2018;9:5416。
Schafer ST、Paquola ACM、Stern S、Gosselin D、Ku M、Pena M、Kuret TJM、Liyanage M、Mansour AA、Jaeger BN 等人。病理性启动导致自闭症患者来源的神经元中发育基因网络的异时性。《自然神经科学》。2019;22:243-255。
Smith AM、LaValle TA、Shinawi M、Ramakrishnan SM、Abel HJ、Hill CA、Kirkland NM、Rettig MP、Helton NM、Heath SE等。DNMT3A过度生长综合征患者和小鼠的功能表型及表观遗传表型。《自然·通讯》,2021;12:4549。
Tatton-Brown K、Loveday C、Yost S、Clarke M、Ramsay E、Zachariou A、Elliott A、Wylie H、Ardissone A、Rittinger O等人。表观遗传调控基因的突变是过度生长伴智力障碍的主要原因。《美国人类遗传学杂志》。2017;100:725-736。
塔顿-布朗 K、默里 A、汉克斯 S、道格拉斯 J、阿姆斯特朗 R、班卡 S、伯德 LM、克莱里库齐奥 CL、科米尔-戴尔 V、库欣 T 等。韦弗综合征与 EZH2 突变:阐明临床表型。《美国医学遗传学杂志 A 卷》。2013;161A:2972-2980。
Tatton-Brown K、Zachariou A、Loveday C、Renwick A、Mahamdallie S、Aksglaede L、Baralle D、Barge-Schaapveld D、Blyth M、Bouma M 等人。《Tatton-Brown-Rahman 综合征:55 例新发组成型 DNMT3A 变异个体的临床研究》。《Wellcome Open Res》。2018;3:46。
托马斯·H、阿历克斯·T、勒纳尔·E、勒诺·M、伍姆斯·J、祖伊·S、勒厄普·B、热纳维耶芙·D、德勒蒙·N、施密特·E等人。24例法国患者中塔顿-布朗-拉赫曼综合征的遗传和临床谱系扩展。《医学遗传学杂志》。2024年。
Ure K、Lu H、Wang W、Ito-Ishida A、Wu Z、He LJ、Sztainberg Y、Chen W、Tang J、Zoghbi HY。在GABA能神经元中恢复Mecp2的表达足以挽救雷特综合征小鼠模型的多种疾病特征。《eLife》。2016;5。
Uzunova G、Pallanti S、Hollander E. 自闭症谱系障碍中的兴奋/抑制失衡:对干预措施和治疗方法的启示。《世界生物精神病学杂志》。2016;17:174-186.
王力,黄丹,蒋志,罗燕,诺里斯·C,张明,田晓,唐勇。Akt3负责胚胎干细胞的存活和增殖。《生物学开放》。2017;6:850–861。
王M、魏PC、林CK、加林娜IS、马歇尔S、马尔凯蒂MC、奥特FW、盖奇FH。自闭症的人诱导多能干细胞模型中神经祖细胞增殖增加导致复制应激相关基因组不稳定性。《细胞干细胞》。2020;26:221–233.e226。
渡边D、内山K、花冈K。神经祖细胞发育过程中小鼠从头甲基转移酶表达从Dnmt3b向Dnmt3a的转变。《神经科学》。2006;142:727-737。
吴Z、黄K、余J、乐T、并平M、刘Y、张J、薛Z、程L、范G。Dnmt3a调节小鼠神经干细胞的增殖和分化。《神经科学研究杂志》。2012;90:1883-1891。
Ziller MJ、Ortega JA、Quinlan KA、Santos DP、Gu H、Martin EJ、Galonska C、Pop R、Maidl S、Di Pardo A 等。解析人类运动神经元分化和生理过程中新发 DNA 甲基化动态的功能后果。《细胞干细胞》。2018;22:559-574.e559。
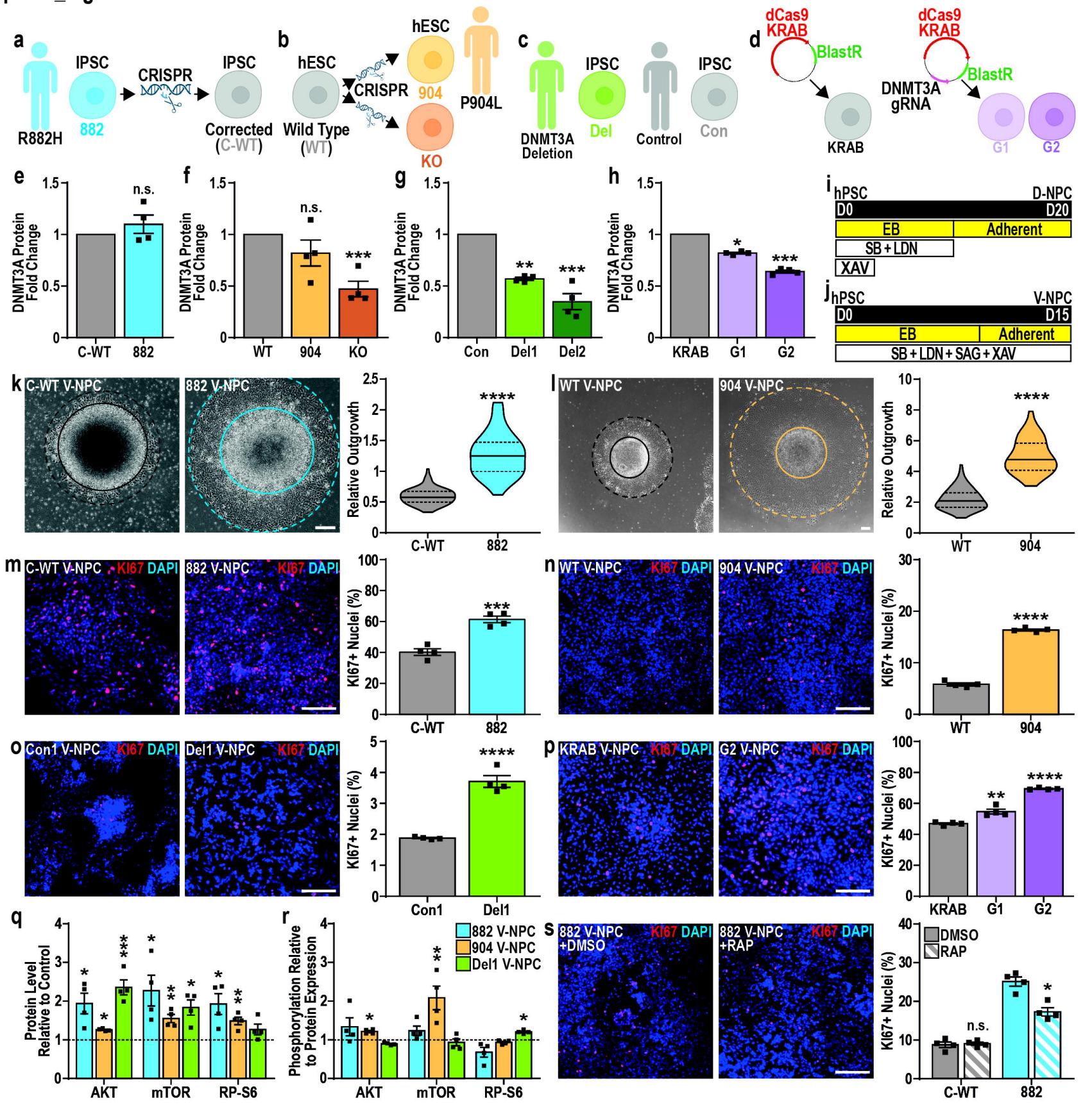
图1. TBRS模型显示V-NPC增殖和mTOR信号传导增强。
a. 携带DNMT3A基因p.R882H突变的诱导多能干细胞(882),来源于一名TBRS患者,以及经校正的同源对照细胞(C-WT)。 b. 对照人胚胎干细胞(WT),通过基因敲入方式进行修饰以模拟携带杂合p.L904M突变的TBRS患者(904),同时构建了DNMT3A两个等位基因均存在小片段(<10 bp)突变的模型(KO)。 c. 从一名携带DNMT3A基因杂合缺失的TBRS患者中生成了两株诱导多能干细胞系(Del1/2),并与性别匹配的同源对照诱导多能干细胞(Con1/2)进行比较。 d、e. 使用两个靶向DNMT3A的向导RNA(G1/2)构建了CRISPRi人胚胎干细胞模型,并与不含向导RNA的人多能干细胞(KRAB)进行比较。 e–h. (e)882、(f)904、(g)KO、(h)Del1/2和(i)G1/2这些TBRS模型与匹配对照中DNMT3A蛋白表达的定量分析。 i–j. (i)D-NPC分化和(j)V-NPC分化的示意图。 k–l. (k)822和(l)904的V-NPC分化第12天时,接种的神经球(粗线)及其相对神经球生长(虚线)的代表性图像和定量分析。 m–p. (m)822、(n)904、(o)Del1/2和(p)G1/2的V-NPC中Ki67免疫阳性的代表性图像和定量分析,与匹配对照相比。 q–r. TBRS的V-NPC(882、904和Del1)与匹配对照(虚线)中AKT、mTOR和核糖体蛋白S6的相对(q)丰度及(r)磷酸化水平的定量分析。 s. 经雷帕霉素(RAP)或溶媒对照(DMSO)处理后,882和C-WT的V-NPC中Ki67阳性的代表性图像和定量分析。
数据表示为平均值±标准误(e–h、m–s)或分布,其中包含中位数(粗线)和上下四分位数(虚线)(k–l),并通过Student t检验与匹配对照组进行比较分析(e–h、k–s)。所有条件下均有4次生物学重复实验,p值:*p<0.05;**p<0.01;***p<0.001;****p<0.0001。比例尺=200 μm(k–l)和100 μm(m–p、s)。
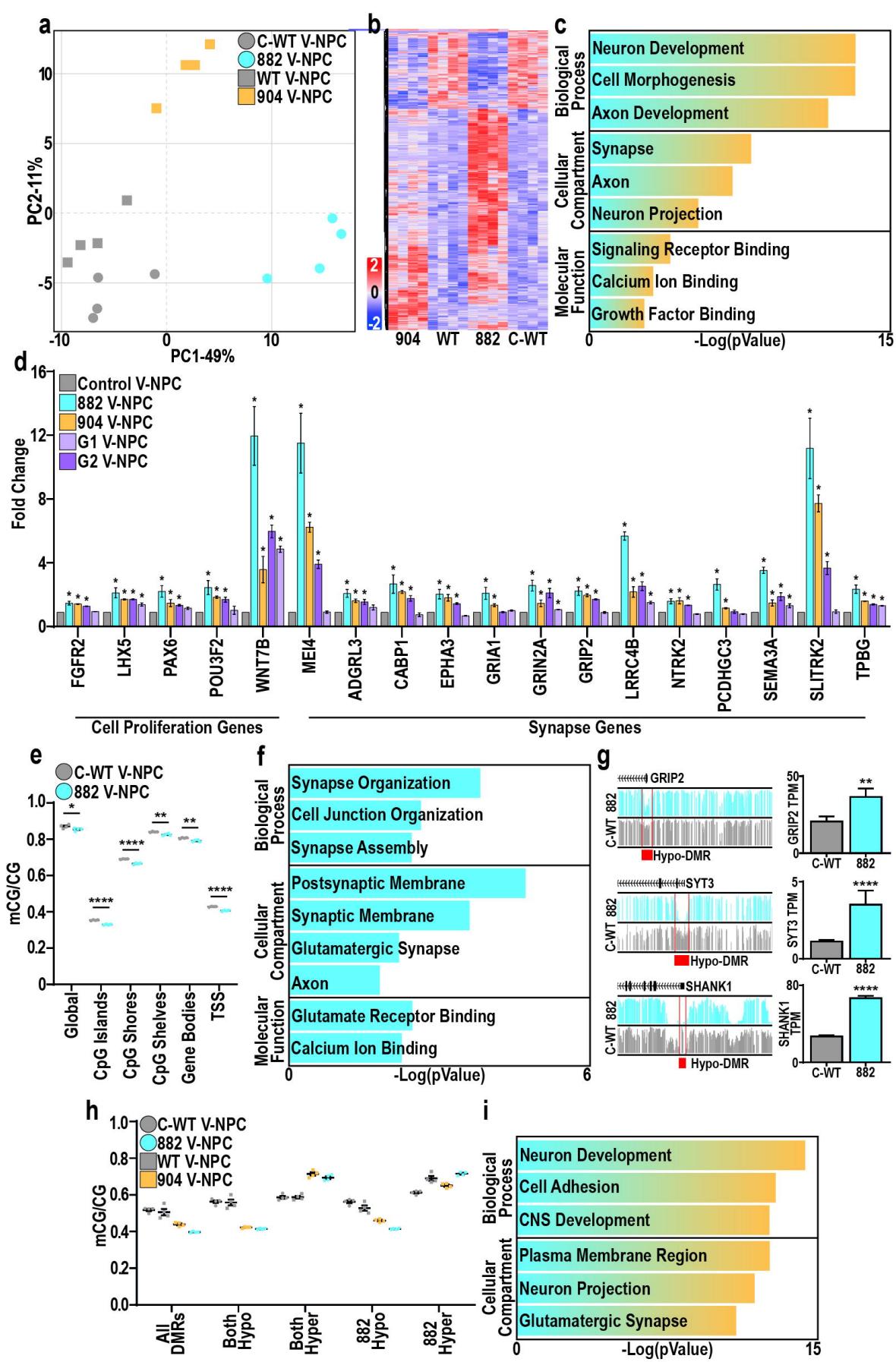
图2. DNMT3A介导V-NPCs中前神经元基因的表观遗传抑制
a. 对照组(灰色)和TBRS组(882-蓝色和904-橙色)V-NPC的主成分分析。 b. 882和904两组相对于对照组(C-WTWT)V-NPC共有的差异表达基因(DEGs)的热图。 c. TBRS组(882/904)相对于对照组V-NPC中上调的差异表达基因的基因本体(GO)富集分析总结。 d. 与“细胞增殖”和“突触”GO术语相关的基因在TBRS组(882、904、G1/2)相对于对照组V-NPC中的表达变化。 e. C-WT组与882组V-NPC中mCG/CG水平的变化。 f. 与882低甲基化差异甲基化区域(hypo-DMR)相关的882差异表达基因的GO富集分析总结。 g. C-WT和882组V-NPC中mCG的基因组浏览器视图,突出显示与882组V-NPC中上调的差异表达基因启动子相关的差异甲基化区域(红色)。 h. 共有差异甲基化区域(全部)的mCG/CG差异,按两个TBRS模型中差异甲基化区域的方向(均高甲基化或均低甲基化)或仅在882模型中(882高甲基化或882低甲基化)进行分类。 i. TBRS组(904和882)V-NPC中上调且与共有低甲基化差异甲基化区域相关的差异表达基因的GO富集分析总结。
数据表示为平均值±标准误(d、e、g、h),并标注了各个生物学重复(e、h)。通过Student t检验与同基因对照进行数据分析(d、e),或通过差异表达分析进行数据分析(g)。所有条件下均有4次生物学重复实验,p值:*p<0.05(d、e);**p<0.01;***p<0.001(e、g)。
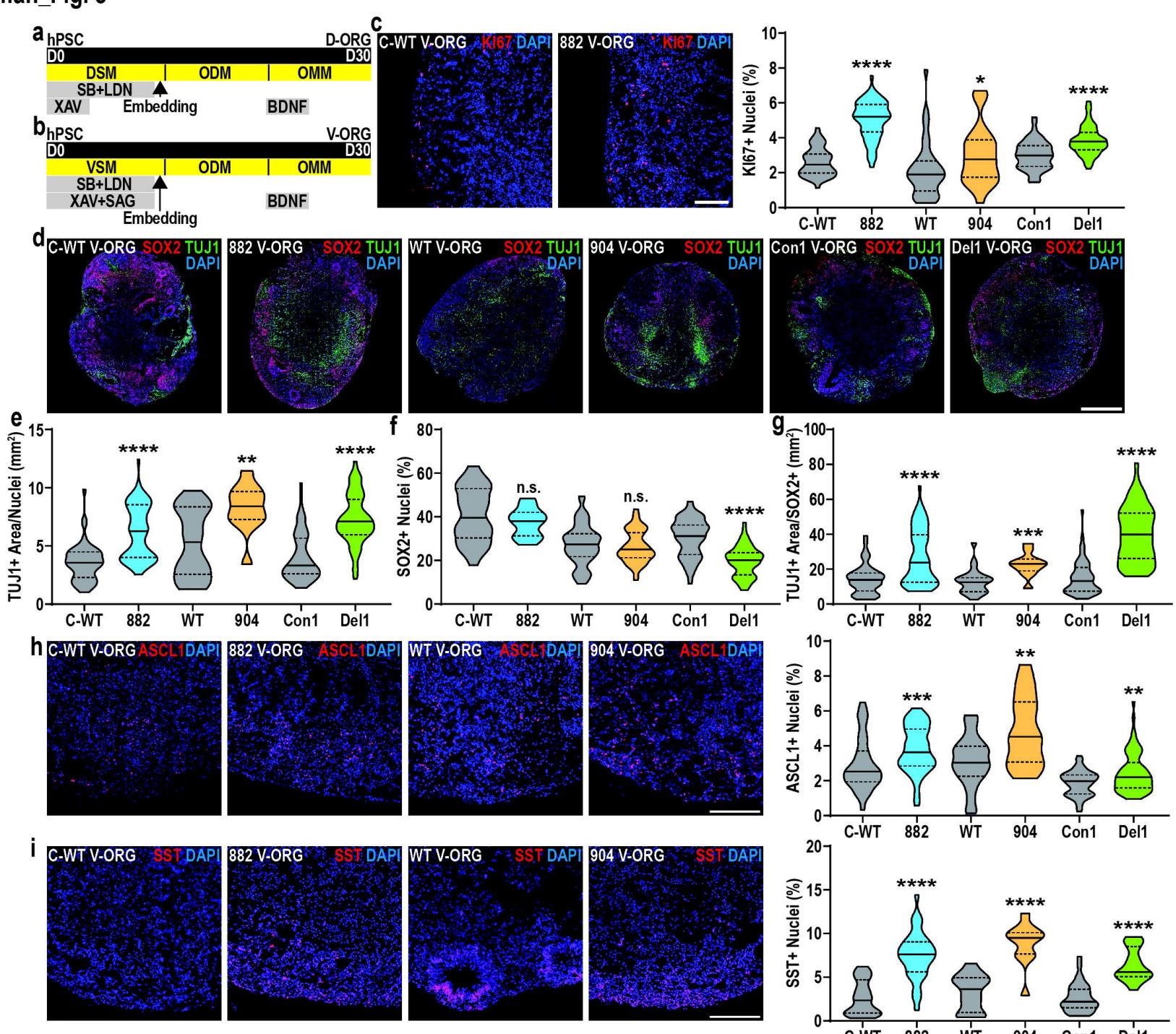
图3. TBRS V-ORGs显示出增殖和神经发生增强
a. (a)D-ORG或(b)V-ORG分化的示意图。c. TBRS组和对照组V-ORG中Ki67+细胞比例的代表性图像及定量分析。d-g. TBRS组和对照组V-ORG中(d)SOX2和TUJ1染色的代表性图像,以及(e)TUJ1+区域、(f)SOX2+比例(占DAPI+细胞核的百分比)和(g)相对于SOX2+细胞标准化的TUJ1+区域的定量分析。h-i. TBRS组和对照组V-ORG中(h)ASCL1+和(i)SST+细胞比例的代表性图像及定量分析。
所有数据均以分布形式呈现,其中中位数(粗线)和上下四分位数(虚线)已标出,并通过曼-惠特尼检验进行分析,将每个TBRS模型与其匹配的对照组进行比较。每种条件至少制备3批类器官,每批评估3-9个类器官。p值:*p<0.05;**p<0.01;***p<0.001;****p<0.0001;n.s.-不显著。比例尺=100μm(c、h-i)和500μm(d)。
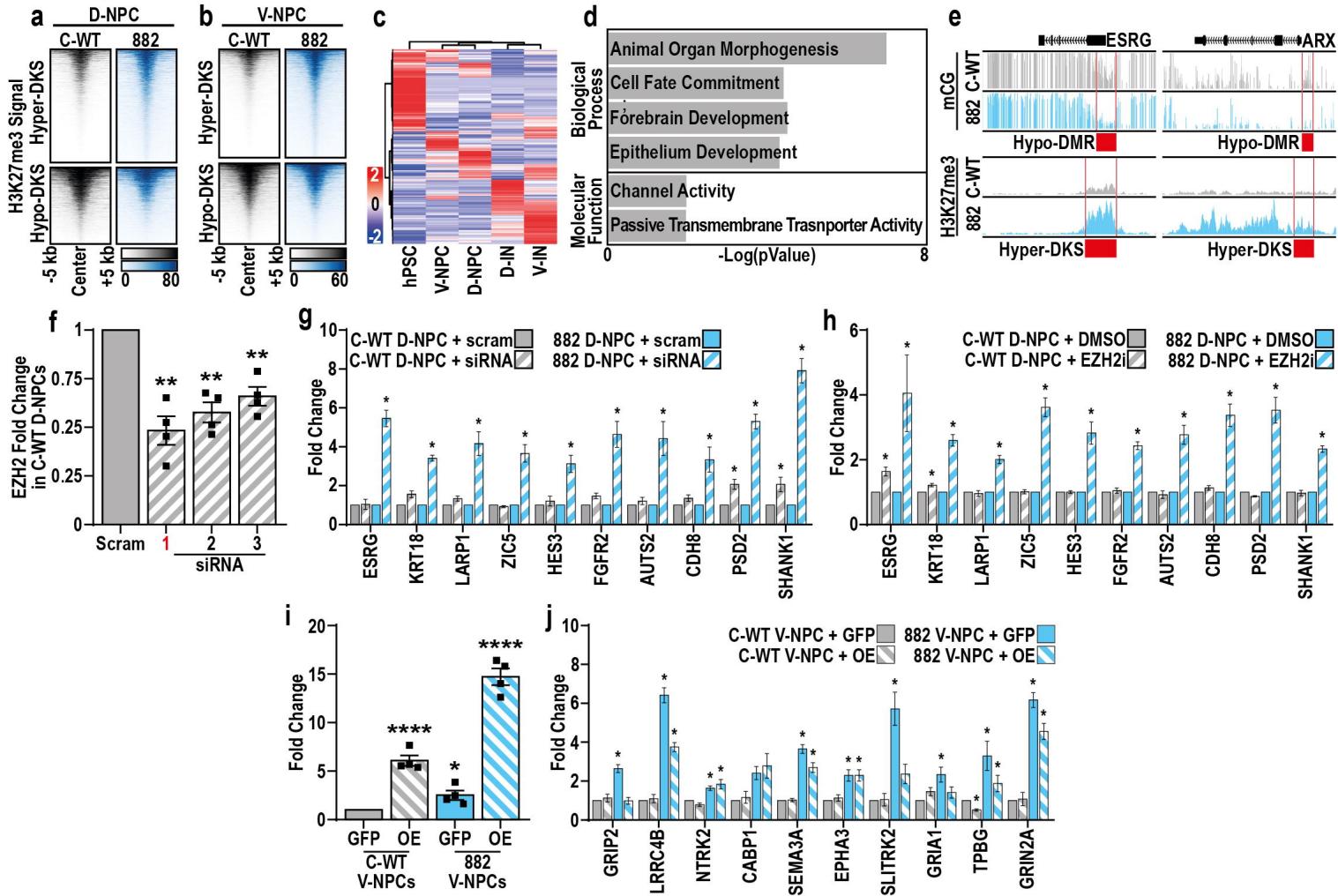
图4. H3K27me3的增加可特异性补偿R882H神经祖细胞中DNA甲基化的缺失
a. 与对照组相比,882组中H3K27me3峰显著增加(高DKS)或减少(低DKS):(a) D-NPCs或(b) V-NPCs。 c. 在神经元分化过程中,882组NPCs(D和V)中与低DMRs和低DKSs相关的基因表达。 d. 882组D-NPCs(D和V)中与低DMRs和低DKSs相关的非差异表达基因的GO富集分析摘要。 e. 882组和对照组V-NPCs中mCG和H3K27me3的基因组浏览器视图,突出显示(红色)与两个基因启动子相关的低DMRs和高DKSs。 f. 用 scramble siRNA(siRNA)或靶向EZH2的siRNA(1-3)处理后,C-WT D-NPCs中EZH2 mRNA水平的定量分析。 g-h. 在用(g)siRNA或(h)EZH2特异性抑制剂处理后,与882组D-NPCs低DMRs和高DKSs相关的10个基因的表达变化定量分析。 i. 慢病毒转导过表达EZH2(OE)或GFP(FIP)的构建体后,C-WT和882组V-NPCs中EZH2 mRNA水平的定量分析。 j. 在OE或GFP条件下,882组V-NPCs中10个上调基因的表达变化定量分析。
数据以平均值±标准误(e-j)表示,并采用单因素方差分析进行分析,将处理组分别与(f、g) scramble对照组、(h)DMSO对照组或(i-j)GFP对照组进行比较。所有条件下均进行了4次生物学重复实验,p值:*p<0.05;**p<0.01;****p<0.0001(f、g、h-j)。
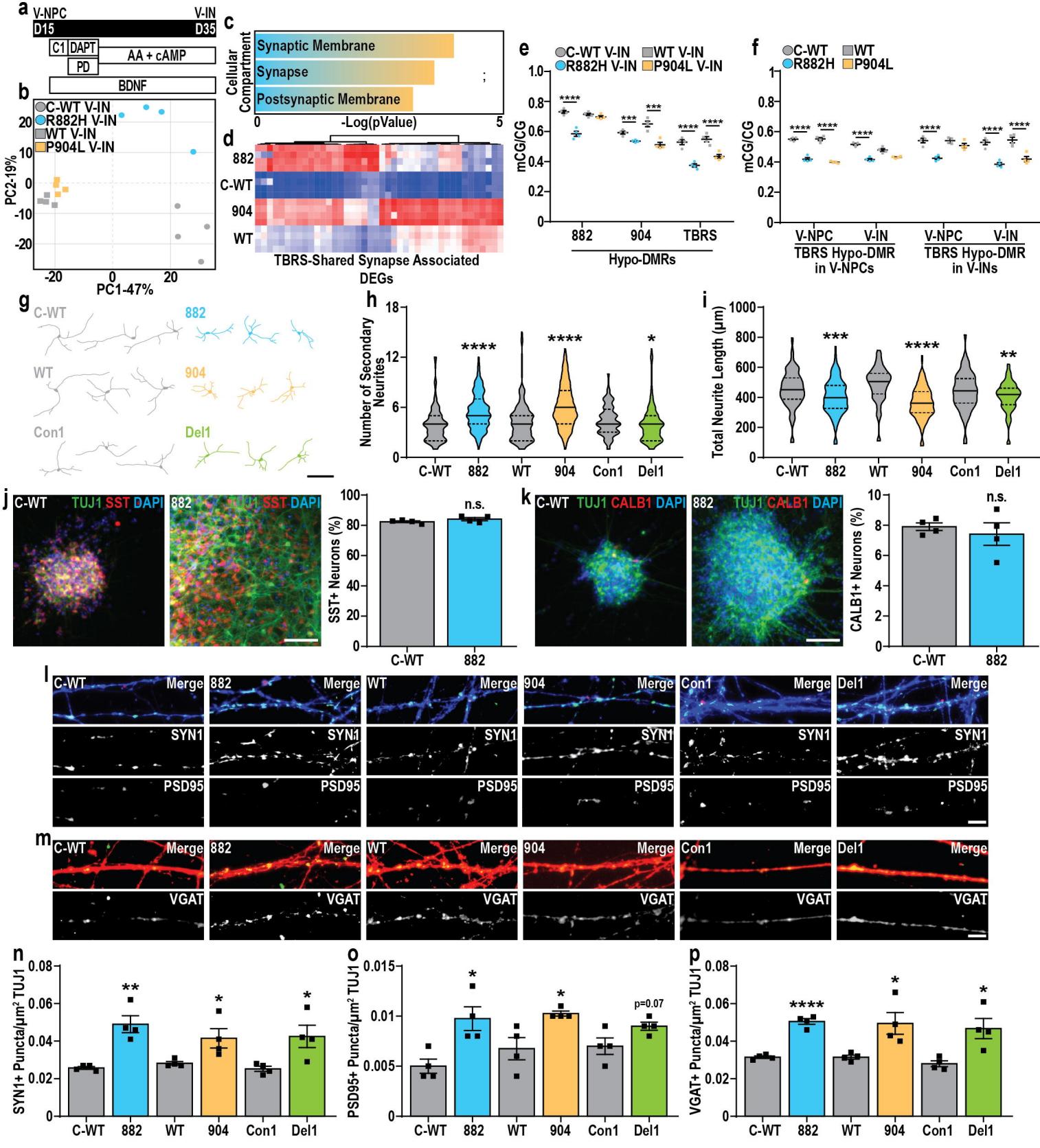
图5. DNMT3A在中间神经元分化过程中抑制神经元成熟
a. 从腹侧模式神经祖细胞(V-NPCs)生成γ-氨基丁酸能中间神经元(V-Ins)的实验范式。 b. 对照组(灰色)和TBRS组(882-蓝色和904-橙色)V-Ins的主成分分析。 c. TBRS V-Ins中上调的差异表达基因(DEGs)的基因本体论(GO)富集分析总结。 d. 与“突触”GO术语相关且在TBRS V-Ins中上调的基因热图。 e. 882、904或TBRS共有的V-IN低甲基化差异甲基化区域(hypo-DMRs)以及匹配的对照V-Ins中的mCG/CG变化。 f. 在TBRS和对照V-NPCs及V-Ins中,V-NPCs或V-Ins中识别到的TBRS D-Ins共有低甲基化差异甲基化区域的mCG/CG变化。 g. 用于量化神经元形态的对照组和TBRS组V-Ins的代表性图像(包括轨迹)。 h-i. 对照组和TBRS组第30天V-Ins的神经元形态量化结果。 j-k. 第40天C-WT或882 V-Ins培养物中(j)生长抑素(SST)或(k)钙结合蛋白1(CALB1)阳性神经元比例的代表性图像和量化结果。 l-m. TBRS组和对照组第50天V-Ins中突触前标志物(SYN1)和突触后标志物(PSD95) puncta密度以及mP-VGAT的代表性图像和量化结果。
采用Student t检验与匹配对照组对数据进行分析(e、f、h、i、n-p)。所有条件下均进行了4次生物学重复实验,每个生物学重复至少对20个神经元进行量化测量。p值:*p<0.05;**p<0.01;***p<0.001;****p<0.0001。比例尺:100 μm(g)、50 μm(j、k)和10 μm(l、m)。
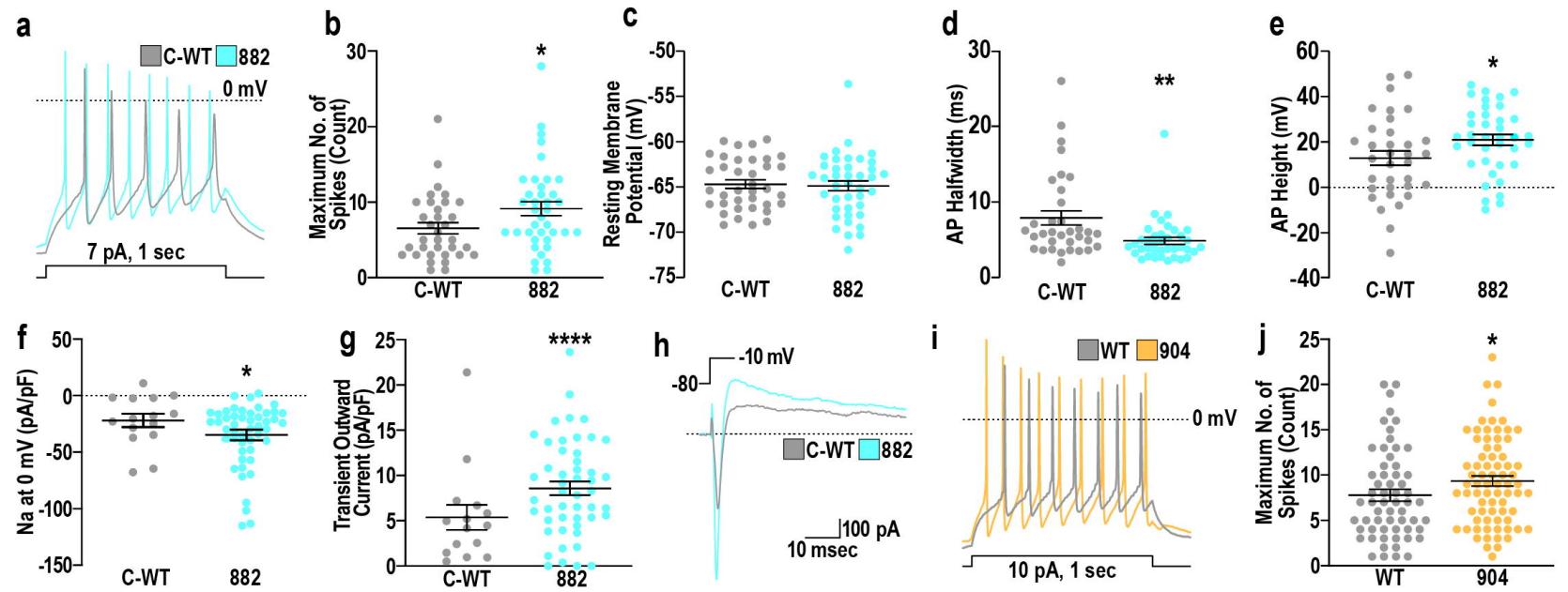
图6. DNMT3A中与TBRS相关的突变导致γ-氨基丁酸能神经元过度活跃
a. 1秒刺激诱导的C-WT(灰色)或882(蓝色)GABA能神经元中动作电位的代表性示例轨迹。 b. 1秒刺激期间C-WT和882 GABA能神经元的最大尖峰数量量化。 c. C-WT和882 GABA能神经元的静息膜电位。 d-e. 动作电位(AP)特征的量化,包括(d)C-WT和882 GABA能神经元的AP半宽和(e)AP高度。 f-g. (f)0 mV时钠(Na)电流密度和(g)电压阶跃后的瞬时外向电流的测量值,均通过电容(pA/pF)标准化。 h. 从-80到-10 mV的电压阶跃期间C-WT和882 GABA能神经元中电流变化的示例轨迹。 i. 1秒刺激诱导的C-WT(灰色)或904(橙色)GABA能神经元中动作电位的代表性示例轨迹。 j. 1秒刺激期间在谷氨酸能和GABA能神经元(合并数据分析)中测量的最大尖峰数量量化。
显著性通过秩和检验计算,结果以平均值±标准误表示,数据点代表从单个神经元获得的测量值,p值:*p<0.05;**p<0.01;***p<0.001。
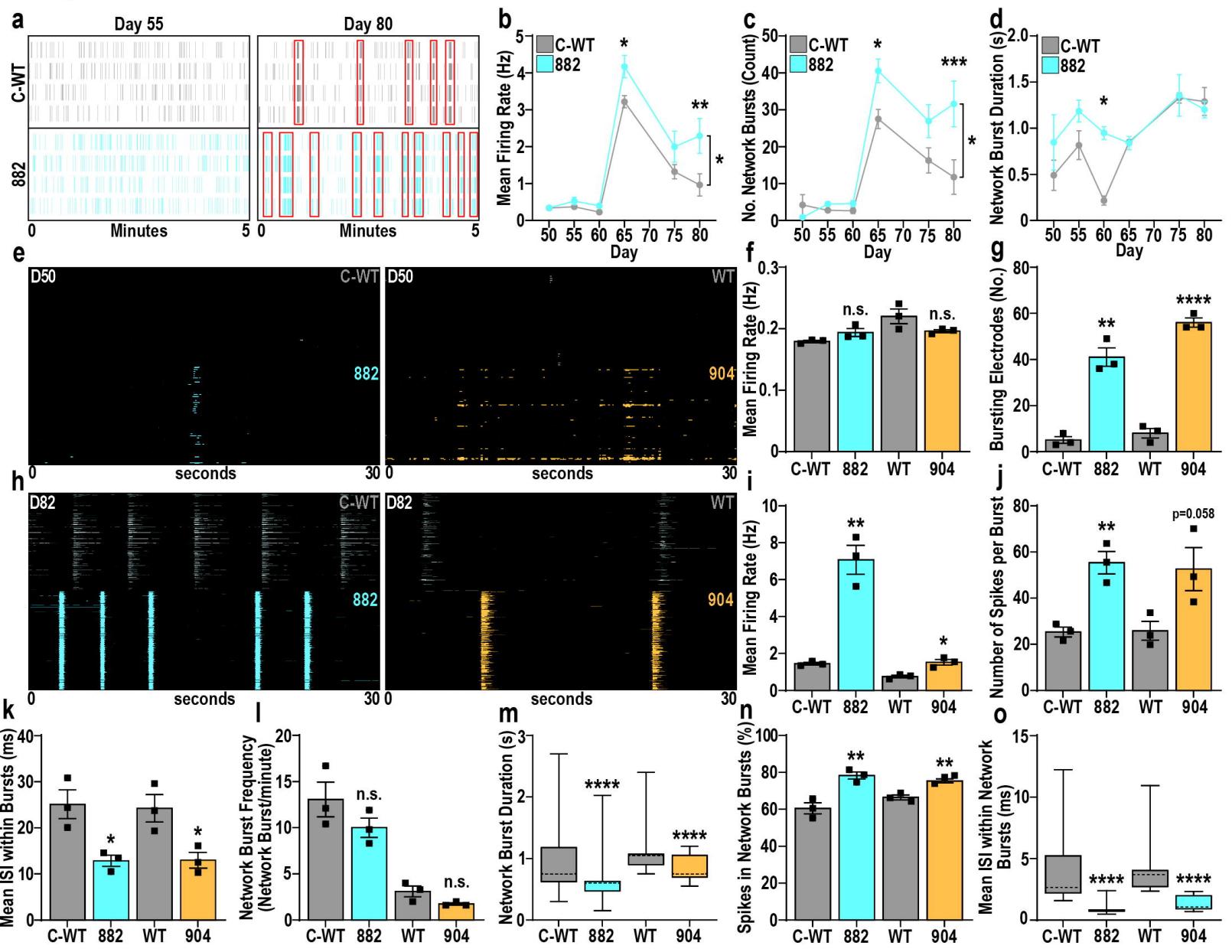
图7. GABA能神经元过度活跃导致TBRS神经元网络功能障碍
a. C-WT或882 GABA能/谷氨酸能神经元共培养物中LD-MEA活性的代表性光栅图,时间点从早期(第55天,左)到晚期(第80天,右),红色方框标示同步活动。b-d. C-WT和882神经元网络的特征,突出显示882神经元网络中(b) firing rate升高(F₁₆=6.579,P<0.05,n=4)和(c)网络爆发频率增加(F₁₁₂=12.18,P<0.0005,n=4),而(d)网络爆发持续时间无显著变化。e-g. HD-MEA活性的代表性光栅图(e),评估分化50天后TBRS GABA能神经元对神经元网络功能障碍的影响,突出显示TBRS GABA能神经元对(f)firing rate和(g)神经元爆发活动出现的影响。h-o. 分化82天后,HD-MEA活性评估TBRS GABA能神经元对神经元网络功能障碍的影响。h-i. 包含TBRS GABA能神经元的培养物中firing rate变化的代表性光栅图和量化结果。j-k. TBRS GABA能神经元改变的神经元爆发参数特征,包括(j)每次爆发的尖峰数量和(k)神经元爆发内尖峰之间的尖峰间隔(ISI)。l-o. 存在TBRS GABA能神经元时的神经元网络参数特征,包括网络爆发(l)频率和(m)持续时间,以及(n)网络爆发内的活动比例和(o)网络爆发内尖峰的ISI。
采用双因素方差分析(ANOVA)对数据进行分析,并进行事后Tukey多重比较检验(b–d)。采用Student t检验(f、g、i、k、l、n)或Mann-Whitney检验(m、o)与同基因对照进行比较。数据以平均值±标准误(SEM)表示(b–d、f、g、i、k、l、n),数据点代表各个生物学重复(f、g、i、k、l、n);或采用箱线图表示,其中虚线标示平均值(m、o)。所有实验均包含3个生物学重复;*P<0.05;**P<0.01;***P<0.001;****P<0.0001;n.s. = 无显著性差异。